19/2007. (III. 29.) FVM rendelet
a Magyar Élelmiszerkönyv közösségi előírások átvételét megvalósító kötelező előírásairól szóló 56/2004. (IV. 24.) FVM rendelet módosításáról1
2009.11.27.
2. § Ez a rendelet 2007. július 1-jén lép hatályba.
Melléklet a 19/2007. (III. 29.) FVM rendelethez4
I.5
II.6
* * *
MAGYAR ÉLELMISZERKÖNYV
(Codex Alimentarius Hungaricus)
3–1–79/1067 számú előírás
(2. kiadás – 2006.)
Sűrített tej és tejporfélék vizsgálata
Ha azonos célra többféle módszer alkalmazható, ezek bármelyikével vizsgálhatunk. Az alkalmazott módszert a 2. számú melléklet szerint elkészített vizsgálati jegyzőkönyvben meg kell adni. Ez az előírás 2007. július 1-jén lép hatályba, ezzel egyidejűleg a Magyar Élelmiszerkönyvnek a sűrített tej és tejporfélék vizsgálatáról szóló 3–1–79/1067 számú előírása 1995-ben jóváhagyott 1. kiadása hatályát veszti.
Ez az előírás az egyes emberi fogyasztásra szánt, részben vagy teljesen dehidratált tartós tejtermékek vizsgálatára szolgáló közösségi elemzési módszerek megállapításáról szóló, 1979. november 13-i 79/1067/EGK bizottsági irányelvnek való megfelelést szolgálja.
1. számú melléklet a 3–1–79/1067 számú előírás 2. kiadásához
Sűrített tej és tejporfélék vizsgálati módszereinek alkalmazási területe
I. Bevezetés, általános előírások
II. Szárazanyag-tartalom meghatározás:
III. Víztartalom meghatározás:
IV. Zsírtartalom meghatározás:
V. Szacharóz meghatározás:
VI. Tejsav és laktát meghatározás:
VII. Foszfatáz-aktivitás meghatározás:
2. számú melléklet a 3–1–79/1067 számú előírás 2. kiadásához
Sűrített tej és tejporfélék vizsgálati módszerei
Bevezetés, általános előírások
1. Mintavétel
1.1. Cukrozatlan sűrített zsíros tej
Cukrozatlan sűrített tej
Cukrozatlan sűrített félzsíros tej
Cukrozatlan sűrített soványtej
A lezárt dobozt összerázzuk és megforgatjuk. A dobozt kinyitjuk és a tejet lassan egy másik, légmentesen zárható edénybe töltjük át, majd ismételt áttöltögetéssel összekeverjük. Gondoskodjunk arról, hogy a doboz falán és alján tapadó zsír- és tejrészecskék a mintába kerüljenek. Az edényt lezárjuk.
Ha a tartalom nem homogén, akkor az edényt 40 °C hőmérsékletű vízfürdőn melegítjük, 15 percenként erősen megrázzuk. 2 óra múlva az edényt a vízfürdőről levesszük, szobahőmérsékletre hűtjük. A fedőt levesszük és az edény tartalmát kanállal vagy spatulával alaposan összekeverjük (amennyiben a zsír kivált, a minta nem vizsgálható). Hűtve tároljuk.
1.2. Cukrozott sűrített tej
Cukrozott sűrített félzsíros tej
Cukrozott sűrített soványtej
Doboz: a lezárt dobozt vízfürdőben 30–40 °C-on kb. 30 percig melegítjük. A dobozt kinyitjuk és tartalmát kanállal vagy spatulával felfelé, lefelé és körkörös irányú mozgatással jól összekeverjük a felső és alsó rétegek elegyedése végett. Gondoskodjunk arról, hogy a doboz karimáján, falán és alján tapadó tejrészecskék a mintába jussanak. A doboz tartalmát amilyen gyorsan csak lehet, légmentesen záró fedéllel ellátott másik dobozba öntjük. Az edényt lezárjuk, és hidegen tároljuk.
Tubus: alját levágjuk, és tartalmát egy légmentesen záró fedéllel ellátott edénybe nyomjuk. Aztán a tubust hosszában is felvágjuk. Belsejéből az összes anyagot kikaparjuk, és a többivel gondosan összekeverjük. Az edényt hűtve tároljuk.
1.3. Emelt zsírtartalmú tejpor
Teljes tejpor
Félzsíros tejpor
Sovány tejpor
A tejport tiszta, száraz, légmentesen záródó fedővel ellátott edénybe töltjük. Az edény térfogata a por térfogatának kétszerese legyen. Az edényt azonnal lezárjuk és a tejport az edény ismételt rázásával és forgatásával alaposan összekeverjük. A minta előkészítése során amennyire csak lehet, kerüljük el, hogy a tejpor a levegővel érintkezzen, s ez által nedvességet vegyen fel.
2. Vegyszerek
2.1. Víz
2.1.1. Minden olyan esetben, amikor oldásra, hígításra vagy mosásra vizet használunk, az mindig desztillált vagy legalább azzal azonos minőségű, ionmentesített víz legyen.
2.1.2. Minden olyan esetben, amikor „oldás” vagy „hígítás” minden további adat nélkül szerepel, mindig „vízben való oldást” vagy „vízzel való hígítást” kell értenünk.
2.2. Tisztasági követelmény
Minden alkalmazott vegyszer analitikai tisztaságú legyen, hacsak nincs más kikötés.
3. Eszközök
3.1. Készülékek és segédanyagok felsorolása
A módszereknél a felsorolás csak a speciális készülékeket és eszközöket tartalmazza.
3.2. Analitikai mérleg, amelynek pontossága legalább 0,1 mg.
4. Az eredmények kiszámítása
4.1. A beltartalom kiszámítása
Ha nincs egyéb előírás, a laboratórium által számított eredményt mindig a minta tömegszázalékában adjuk meg.
4.2. Az eredmény megadásának pontossága
Az eredményben nem szabad több tizedesjegyet megadni mint amennyit az alkalmazott analitikai módszer pontossága megenged.
5. Vizsgálati jegyzőkönyv
A vizsgálati jegyzőkönyvben az analitikai módszereket és az eredményeket kell megadni. Kiegészítésképpen adjunk meg minden olyan részletet, amelyet a vizsgálati módszer nem ír elő vagy választható és minden olyan körülményt, amely az eredményt befolyásolhatja.
A vizsgálati jegyzőkönyv tartalmazza a minta azonosításához szükséges összes adatot.
1. módszer: Szárazanyag-tartalom meghatározás (szárítószekrényben 99 °C-on)
1. Tárgy és alkalmazási terület
Ezzel a módszerrel a következő termékek szárazanyagtartalmát határozzuk meg:
– cukrozatlan sűrített zsíros tej,
– cukrozatlan sűrített tej,
– cukrozatlan sűrített félzsíros tej,
– cukrozatlan sűrített soványtej,
– cukrozott sűrített tej,
– cukrozott sűrített félzsíros tej,
– cukrozott sűrített soványtej.
2. Fogalommeghatározás
A sűrített tej szárazanyaga: az ezzel a módszerrel meghatározott szárazanyag.
3. A módszer elve
A minta ismert mennyiségét vízzel hígítjuk, homokkal elkeverjük és 99±1 °C-on szárítjuk. A szárítás utáni tömeg a szárazanyag. A szárazanyagot a minta tömegszázalékában adjuk meg.
4. Vegyszerek és segédanyagok
Kvarchomok vagy tengeri homok, sósavval kezelt (szemcsemérete: 0,18–0,5 mm, ez azt jelenti, hogy 500 mikronos szitaszöveten átesik, de 180 mikronos szitán fennmarad). Végezzük el a következő vizsgálatot:
Körülbelül 25 g homokot 2 órán keresztül szárítószekrényben (5.3.) a 6.1. és 6.3. pontokban leírtak szerint szárítsunk. Adjunk hozzá 5 ml vizet és szárítószekrényben 2 órán keresztül ismét szárítsuk, majd hűtsük le és még egyszer mérjük le. A két tömeg közötti különbség 0,5 mg-nál nem lehet több.
Ha szükséges, a homokot sósavoldattal (25%) 3 napig kezeljük, időnként megkeverjük. Vízzel mossuk sav-, illetve kloridmentesre. 160 °C-on szárítsuk, és a fentiek szerint újra vizsgáljuk meg.
5. Eszközök
5.1. Analitikai mérleg
5.2. Bemérőedény fémből, nikkelből, alumíniumból, rozsdamentes acélból vagy üvegből. A bemérőedénynek jól záró, de gyorsan levehető fedele legyen.
Megfelelő méretek: átmérő 60–80 mm, mélység kb. 25 mm.
5.3. Szárítószekrény, atmoszférikus nyomású, jól szellőző, 99 °C±1 °C-ra beállítható. A hőmérséklet az egész szekrényben azonos legyen.
5.4. Exszikkátor, nedvességindikátort tartalmazó, frissen aktívált szilikagéllel vagy azonos hatékonyságú szárító közeggel.
5.5. Üvegbot, az egyik végén lapos és olyan hosszú, hogy a bemérőedénybe (5.2.) beleférjen.
5.6. Vízfürdő, forrásban tartható.
6. A vizsgálat menete
6.1. Körülbelül 25 g homokot (4) és egy rövid üvegbotot (5.5.) a bemérőedénybe (5.2.) viszünk.
6.2. Az edényt (levett fedővel), a fedőt és a bemért homokot 2 órán keresztül szárítószekrényben (5.3.) szárítjuk.
6.3. A fedőt újra rátesszük, és a bemérőedényt az exszikkátorba helyezzük. Szobahőmérsékletre hűtjük és 0,1 mg pontossággal mérjük. (Mo)
6.4. A homokot az edény egyik oldalára döntjük. A szabad térbe a cukrozott sűrített tejből kb. 1,5 g-ot, a cukrozatlan sűrített tejből 3,0 g-ot adagolunk. A fedőt rátesszük, és 0,1 mg pontossággal lemérjük. (M1)
6.5. A fedőt levesszük, 5 ml vizet adunk hozzá és az üvegbot (5.5.) segítségével keverjük össze előbb a folyadékokat, majd a homokot és a folyékony részt. A botot a keverékben hagyjuk.
6.6. A bemérőedényt forrásban lévő vízfürdőn (5.6.) tartjuk, amíg a víz el nem párolog. Ez általában 20 percig tart. A keveréket időnként üvegbottal megkeverjük, hogy az anyagot jól átszellőztessük, és hogy a szárítás alatt pogácsává ne álljon össze. Az üvegbotot a bemérőedényben hagyjuk.
6.7. Az edényt és a fedőt 1 óra 30 percre szárítószekrénybe helyezzük.
6.8. A fedőt újra rátesszük, és az edényt exszikkátorba (5.4.) helyezzük, szobahőmérsékletre hűtjük, majd 0,1 mg pontossággal lemérjük.
6.9. Az edényt kinyitjuk és fedelével együtt 1 órán át szárítószekrényben szárítjuk.
6.10. A munkafolyamatot a 6.8. pontban leírtak szerint ismételjük.
6.11. A 6.9. és 6.10. pontban leírt munkafolyamatot addig ismételjük, amíg két egymás utáni mérés közötti tömegkülönbség 0,5 mg-nál kisebb vagy a tömeg nőni nem kezd. Tömegnövekedés esetén a számolásnál (7.) a legalacsonyabb tömegértéket vesszük figyelembe. A végleges tömeget M2-vel jelöljük.
7. Az eredmények kiszámítása
A minta tömegszázalékában megadott szárazanyag-tartalmát a következő képlettel számítjuk ki:
ahol:
M0 = a bemérőedény, az üvegbot, a fedő és a homok együttes tömege g-ban a 6.3. művelet után,
M1 = a bemérőedény, az üvegbot, a fedő, a homok és a minta tömege g-ban a 6.4. művelet után,
M2 = a bemérőedény, az üvegbot, a fedő, a homok és a kiszárított minta tömege g-ban a 6.11. művelet után.
7.3. A cukrozatlan sűrített tej zsírmentes szárazanyagát a következőképpen kapjuk meg: az összes szárazanyagból (2. számú melléklet, 1. módszer) levonjuk a zsírtartalmat (2. számú melléklet, 3. módszer). 8. Ismételhetőség
Párhuzamos vizsgálatok között, azaz ugyanabból a mintából, ugyanazon vizsgáló által egy időben vagy közvetlenül egymás után végzett vizsgálatok eredményei között a megengedett eltérés legfeljebb 0,2 g szárazanyag/100 g termék lehet.
2. módszer: Víztartalom meghatározás (szárítószekrényben 102 °C-on)
1. Tárgy és alkalmazási terület
Ezzel a módszerrel határozzuk meg a következő termékek víztartalmát:
– emelt zsírtartalmú tejpor,
– teljes tejpor,
– félzsíros tejpor,
– sovány tejpor.
2. Fogalommeghatározás
Víztartalom: ezzel a módszerrel meghatározott, a szárítás során bekövetkezett tömegveszteség.
3. A módszer elve
Szárítószekrényben 102±1 °C-on, atmoszférikus nyomáson, tömegállandóságig szárított minta tömegét határozzuk meg. A tömegveszteséget a minta tömegszázalékában számítjuk.
4. Eszközök
4.1. Analitikai mérleg
4.2. Bemérőedény nikkelből, alumíniumból, rozsdamentes acélból vagy üvegből. Az edénynek jól záró, de gyorsan levehető fedele legyen. Megfelelő méretek: átmérő: 60–80 mm, mélység: kb. 25 mm.
4.3. Szárítószekrény, atmoszférikus nyomású, jól szellőző, 102±1 °C-ra beállítható. A hőmérséklet az egész szekrényben azonos legyen.
4.4. Exszikkátor, nedvességindikátort tartalmazó, frissen aktívált szilikagéllel vagy azzal azonos hatékonyságú szárítóanyaggal.
5. A vizsgálat menete
5.1. A bemérőedényt (4.2.) levett fedővel körülbelül 1 órára szárítószekrénybe (4.3.) tesszük.
5.2. Az edényre a fedelet rátesszük és exszikkátorba (4.4.) helyezzük, szobahőmérsékletre hűtjük, és 0,1 mg pontossággal megmérjük. (M0)
5.3. Körülbelül 2 g tejpormintát az edénybe teszünk, az edényt a fedelével lefedjük, és 0,1 mg pontossággal gyorsan lemérjük. (M1)
5.4. Az edényt levett fedővel 2 órára szárítószekrénybe helyezzük.
5.5. Az edényt lefedjük, exszikkátorba helyezzük, és szobahőmérsékletre hűtjük. Lehülés után 0,1 mg pontossággal gyorsan lemérjük. (M2)
5.6. Az edényt levett fedővel 1 órán át szárítószekrényben szárítjuk.
5.7. Az 5.5. pont alatt leírt műveletet megismételjük.
5.8. Az 5.6. és 5.7. szerinti műveleteket addig ismételjük, amíg a két egymás utáni mérés tömege közötti különbség 0,5 mg-nál kisebb vagy a tömeg nőni nem kezd. Ha tömegnövekedés lép fel, a számolásnál a legkisebb tömegértéket vesszük figyelembe (6.). A végleges tömeget M2-vel jelöljük.
6. Az eredmények kiszámítása
A minta tömegveszteségét tömegszázalékban kifejezve a következő képlettel számítjuk ki:
ahol:
M0 = a bemérőedény és a fedél tömege g-ban az 5.2. művelet után,
M1 = a bemérőedény, a fedél, a minta tömege g-ban az 5.3. művelet után,
M2 = a bemérőedény, a fedél és a kiszárított minta tömege g-ban az 5.5. művelet után.
7. Ismételhetőség
Párhuzamos vizsgálatok között, azaz ugyanazon a mintából, ugyanazon vizsgáló által egyidejüleg vagy közvetlenül egymás után végzett vizsgálatok eredményei között a megengedett eltérés legfeljebb 0,1 g víz/100 g termék lehet.
3. módszer: Zsírtartalom meghatározás sűrített tejből (Röse–Gottlieb-módszer)
1. Tárgy és alkalmazási terület
E módszerrel határozzuk meg a zsírtartalmat a következő termékekben:
– cukrozatlan sűrített zsíros tej,
– cukrozatlan sűrített tej,
– cukrozatlan sűrített félzsíros tej,
– cukrozatlan sűrített soványtej,
– cukrozott sűrített tej,
– cukrozott sűrített félzsíros tej,
– cukrozott sűrített soványtej.
2. Fogalommeghatározás
A sűrített tej zsírtartalma: a megadott módszerrel meghatározott zsírtartalom.
3. A módszer elve
A zsírtartalmat Röse–Gottlieb-módszerrel határozzuk meg. A minta ammóniás-alkoholos oldatából a zsírokat dietil-éterrel vagy petroléterrel extraháljuk, majd az oldatot bepároljuk, a maradékot mérjük, és a zsírtartalmat a minta tömegszázalékában fejezzük ki.
4. Vegyszerek
Minden reagensnek meg kell felelnie a vakpróbára vonatkozó részben (6.1.) megadott feltételeknek.
Ha szükséges, a reagenseket 1 g vajzsír/100 ml oldószer jelenlétében újra desztilláljuk.
4.1. Ammónia-oldat, kb. 25% (m/m) NH3 tartalommal, sűrűsége 20 °C-on kb. 0,91 g/ml (vagy ismert koncentrációjú, töményebb oldat).
4.2. Etanol, 96±2%-os (v/v) vagy metanollal, metil-etil-ketonnal, illetve petroléterrel denaturált etanol.
4.3. Dietiléter, peroxidmentes
1. megjegyzés:
A peroxid vizsgálatához 10 ml dietil-étert öntünk üvegdugóval lezárható, kis mérőhengerbe, amelyet előzetesen kevés éterrel átöblítettünk. Hozzáadunk 1 ml frissen készített 10%-os kálium-jodid oldatot, összerázzuk és 1 percig állni hagyjuk. Egyik fázisban sem lehet sárga elszíneződés.
2. megjegyzés:
A dietil-éter peroxidmentesen eltartható, ha olyan nedves cinkfóliát adunk hozzá, amelyet előzőleg 1 percre híg, savas réz-szulfátoldatba merítettünk, majd vízzel leöblítettünk. 1 liter dietil-éterhez kb. 8000 mm2 felületű, hosszú csíkokra felvágott cinkfóliát használjunk. A csíkok legalább az edény feléig érjenek.
4.4. Petroléter, 30–60 °C közötti forrpontú
4.5. Oldószer elegy, közvetlenül a felhasználás előtt azonos térfogatú dietil-éterből (4.3.) és petroléterből készítjük (4.4.). Az oldószerelegy dietil-éterrel vagy petroléterrel helyettesíthető.
5. Eszközök
5.1. Analitikai mérleg
5.2. Megfelelő extrahálócső vagy -lombik, becsiszolt üvegdugóval vagy egyéb, az alkalmazott oldószernek megfelelő zárással.
5.3. Vékonyfalú állólombikok, 150–250 ml névleges térfogatúak.
5.4. Szárítószekrény atmoszférikus nyomású, jól szellőző, hőmérséklete 102±1 °C-ra beállítható.
5.5. Forrást segítő anyag, zsírmentes, nem porózus, használat közben nem törékeny, például üveggyöngy vagy szilícium-karbid darabkák (lásd 6.2.1. pont).
5.6. Pipetta, az extraháló csőhöz megfelelő.
5.7. Centrifuga
6. A vizsgálat menete
6.1. Vakpróba
A minta zsírtartalmának meghatározásával egy időben vakpróbát végzünk 10 ml desztillált vízzel, azonos extraháló készülékben, a reagensek azonos arányával és azonos módon a következőkben leírtak szerint, a 6.2.2. alatt leírtak kivételével. Ha a vakpróbára 0,5 mg-nál nagyobb értéket kaptunk, a reagenseket meg kell vizsgálni, tiszta reagensekkel kell helyettesíteni vagy meg kell tisztítani.
6.2. Vizsgálat
6.2.1. A lombikot (5.3.) az oldószer elpárologtatása során a forrást megkönnyítő anyaggal együtt (5.5.) szárítószekrénybe (5.4.) tesszük, és fél-egy óra hosszat szárítjuk. A lombikot a mérlegszoba hőmérsékletére hűtjük és 0,1 mg pontossággal mérjük.
6.2.2. Az előkészített mintát megkeverjük, és a cukrozott sűrített tej mintákból 2–2,5 g-ot, a cukrozatlan sűrített tej mintákból 4–5 g-ot közvetlen beméréssel vagy visszaméréssel 0,1 mg pontossággal az extraháló edénybe mérünk. 10,5 ml térfogatig vízzel feltöltjük, és enyhe melegítés (40–50 °C) közben az anyagok teljes elkeveredése céljából óvatosan összerázzuk. A minta teljesen eloszlatható legyen, különben a meghatározást meg kell ismételni.
6.2.3. 1,5 ml ammónia-oldatot (4.1.) vagy töményebb oldat egyenértékű térfogatát adunk hozzá és jól összekeverjük.
6.2.4. 10 ml etanolt (4.2.) adunk hozzá és a folyadékokat a nyitott lombikban lassan, tökéletesen összekeverjük.
6.2.5. 25 ml dietil-étert adunk hozzá (4.3.) és folyó vízzel lehűtjük. Az edényt lezárjuk, 1 percig erősen rázzuk és közben többször megfordítjuk.
6.2.6. A dugót óvatosan kivesszük, és 25 ml petrolétert (4.4.) adunk hozzá úgy, hogy a petroléter első néhány milliliterével leöblítjük a dugót és az edény nyakának belső falát. Az öblítőfolyadékot az edénybe engedjük. Az edényt a dugóval újra lezárjuk, 30 másodpercig rázzuk és forgatjuk. Ha a 6.2.7. pontban leírt műveletben nem centrifugálunk, nem szabad túl erősen rázni.
6.2.7. A vizsgálati anyagot a felső folyadékfázis kitisztulásáig, és a vizes fázistól való teljes elválásáig állni hagyjuk. A fázisok szétválasztása megfelelő centrifugával (5.7.) is elvégezhető.
Megjegyzés:
Az a centrifuga, amelyik nem háromfázisú motorral működik, szikrát kelthet. Ezért különösen ügyelni kell arra, hogy robbanás vagy tűz ne keletkezzék az étergőzök jelenléte miatt (például a lombik törése esetén).
6.2.8. A dugót kivesszük, néhány ml oldószer eleggyel a dugót és a lombik nyakának belső falát leöblítjük, az öblítőfolyadékot az extraháló edénybe engedjük. A felső réteget dekantálással vagy pipettával gondosan és lehetőleg teljes mennyiségében a lombikba visszük át (6.2.1.).
Megjegyzés:
Ha a leszivatást nem pipettával végezzük, szükség lehet arra, hogy valamennyi vizet hozzáadva a két folyadék közötti fázishatárt a dekantálás megkönnyítésére megemeljük.
6.2.9. Az extraháló edény nyakának külső és belső falát vagy a pipetta csúcsát és alsó részét néhány ml oldószer eleggyel leöblítjük. Az edény külső oldaláról az öblítőfolyadékot a lombikba, a pipetta és a belső fal öblítőfolyadékát pedig az extraháló edénybe folyatjuk.
6.2.10. A 6.2.5–6.2.9. műveletek megismétlésével 15 ml dietil-éter és 15 ml petroléter felhasználásával második extrakciót végzünk.
6.2.11. Harmadik extrakciót végzünk a 6.2.10. szerint, azonban az utolsó öblítést (6.2.9.) elhagyjuk.
Megjegyzés:
A cukrozatlan és cukrozott sűrített soványtej esetében a harmadik extrakció nem szükséges.
6.2.12. Az oldószert (beleértve az etanolt is) gondos elpárologtatással vagy desztillálással eltávolítjuk. Ha a lombik űrtartalma kicsi, akkor minden extrakció után a fent leírt módon a kis mennyiségű oldószert eltávolítjuk.
6.2.13. Ha már nem érzünk oldószerszagot, a lombikokat fektetve 1 órára a szárítószekrénybe helyezzük és szárítjuk.
6.2.14. A lombikot a mérlegszoba hőmérsékletére hűtjük és 0,1 mg pontossággal mérjük.
6.2.15. A 6.2.13. és a 6.2.14. műveleteket addig ismételjük, míg a két egymás utáni mérés különbsége 0,5 mg-nál kisebb lesz vagy a tömeg növekszik. Tömegnövekedés esetén a számolásnál (7.) a legkisebb értéket vesszük figyelembe. A végső tömeget M1-gyel jelöljük.
6.2.16. Ezután 15–25 ml petrolétert adunk hozzá annak vizsgálatára, hogy az extrahált anyag teljesen oldható-e. Az oldószert enyhén melegítjük és rázogatjuk, amíg a zsír egészen fel nem oldódik.
6.2.16.1. Ha az extrahált anyag petroléterben teljesen oldható, a 6.2.1. és a 6.2.15. mérések közti különbség a zsír tömege.
6.2.16.2. Kétség esetén, vagy ha oldhatatlan anyagok vannak jelen, a lombikban lévő zsírt meleg petroléteres mosással ismét extraháljuk, és az oldhatatlan részt minden dekantálás előtt ülepítjük. A lombik nyakának külső oldalát háromszor leöblítjük. A lombikot 1 órán keresztül fekvő helyzetben szárítószekrényben szárítjuk a 6.2.1. szerint, a mérlegszoba hőmérsékletére hűtjük, majd 0,1 mg pontossággal mérjük.
A zsír mennyisége a 6.2.15. szerinti mennyiségnek és ennek a tényleges végső mennyiségnek a különbsége.
7. Az eredmények kiszámítása
Az extrahált zsír grammban kifejezett tömegét a következő képlettel számítjuk ki:
(M1 – M2) – (B1 – B2)
A minta zsírtartalma %-ban:
|
(M1–M2)– (B1 – B2) |
⋅100 |
|
S |
ahol:
M1 = a lombik tömege zsírral együtt g-ban a 6.2.15. művelet után,
M2 = a lombik tömege g-ban a 6.2.1. művelet után, illetve oldhatatlan anyag jelenléte vagy annak gyanúja esetében a 6.2.16.2. művelet után,
B1 = a vakpróba lombikjának tömege g-ban a 6.2.15. művelet után,
B2 = a vakpróba lombikjának tömege g-ban a 6.2.1. művelet után, illetve oldhatatlan anyag jelenléte vagy annak gyanúja esetén a 6.2.16.2. művelet után,
S = a bemért minta mennyisége g-ban.
8. Ismételhetőség
A párhuzamos vizsgálatok eredményei között, azaz ugyanazon vizsgáló által, ugyanabból a mintából egyidejűleg vagy közvetlenül egymás után végzett vizsgálati eredmények között a megengedett eltérés legfeljebb 0,05 g zsír/100 g termék lehet.
4. módszer: Zsírtartalom meghatározás tejporból (Röse–Gottlieb-módszer)
1. Tárgy és alkalmazási terület
Ezzel a módszerrel határozzuk meg a zsírtartalmat a következő termékekből:
– emelt zsírtartalmú tejpor,
– teljes tejpor,
– félzsíros tejpor,
– sovány tejpor.
2. Fogalommeghatározás
A tejpor zsírtartalma: a megadott módszerrel meghatározott zsírtartalom.
3. A módszer elve
A meghatározást a Röse–Gottlieb-módszer szerint végezzük. A tejpor ammóniás-alkoholos oldatából a zsírt dietil-éterrel és petroléterrel extraháljuk, majd az oldószert elpárologtatjuk, a maradékot mérjük, és a minta tömegszázalékában fejezzük ki.
4. Vegyszerek
Minden reagensnek meg kell felelnie a vakpróbára vonatkozó részben (6.1.) megadott követelményeknek.
Ha szükséges, a reagenseket 1 g vajzsír/100 ml oldószer jelenlétében újra desztilláljuk.
4.1. Ammónia-oldat, kb. 25%-os (m/m) NH3 tartalommal sűrűsége 20 °C-on kb. 0,91 g/ml (vagy ismert koncentrációjú töményebb oldat).
4.2. Etanol, 96±2%-os (v/v) vagy metanollal, metil-etil-ketonnal, illetve petroléterrel denaturált etanol.
4.3. Dietil-éter, peroxidmentes
1. megjegyzés:
A peroxid vizsgálatához 10 ml dietil-étert öntünk üvegdugóval lezárható kis mérőhengerbe, amelyet előzetesen kevés éterrel öblítettük. Hozzáadunk 1 ml frissen készített 10%-os kálium-jodid-oldatot, összerázzuk és 1 percig állni hagyjuk. Egyik fázisban sem lehet sárga elszíneződés.
2. megjegyzés.
A dietil-éter peroxidmentesen eltartható, ha olyan nedves cinkfóliát adunk hozzá, amelyet előzőleg 1 percre savas réz-szulfát-oldatba merítettük, majd vízzel leöblítettük.
Egy liter dietil-éterhez kb. 8000 mm2 felületű, csíkokra felvágott cinkfóliát használjunk. A csíkok legalább az edény feléig érjenek.
4.4. Petroléter, 30–60 °C közötti forrpontú
4.5. Oldószer elegy, melyet közvetlenül a felhasználás előtt azonos térfogatú dietil-éterből (4.3.) és petroléterből (4.4.) készítünk. Az oldószerelegy dietil-éterrel vagy petroléterrel helyettesíthető.
5. Eszközök
5.1. Analitikai mérleg
5.2. Megfelelő extrahálócső vagy lombik becsiszolt üvegdugóval vagy egyéb, az alkalmazott oldószernek megfelelő zárással.
5.3. Vékonyfalú állólombik, 150–250 ml névleges térfogatú.
5.4. Szárítószekrény, atmoszférikus nyomású, jól szellőző, 102±1 °C-ra beállítható.
5.5. Forrást segítő anyag, zsírmentes, nem porózus, használat közben nem törékeny, például üveggyöngy vagy szilícium-karbid darabkák (lásd a 6.2.1. pont).
5.6. Vízfürdő, 60–70 °C-ra beállítható.
5.7. Pipetta, az extraháló csőhöz megfelelő.
5.8. Centrifuga.
6. A vizsgálat menete
6.1. Vakpróba
A minta zsírtartalmának meghatározásával egyidőben vakpróbát végzünk 10 ml desztillált vízzel, azonos extraháló edényben, a reagensek azonos arányával és azonos módon, a következőkben leírtak szerint, a 6.2.2. alatt leírtak kivételével. Ha a vakpróbára 0,5 mg-nál nagyobb értéket kapunk, a reagenseket meg kell vizsgálni, tiszta reagensekkel kell helyettesíteni vagy meg kell tisztítani.
6.2. Vizsgálat
6.2.1. A lombikot (5.3.) az oldószer elpárologtatása során a forrást megkönnyítő anyaggal együtt szárítószekrénybe (5.4.) tesszük és fél-egy óra hosszat szárítjuk. A lombikot a mérlegszoba hőmérsékletére hűtjük, és 0,1 mg pontossággal mérjük.
6.2.2. Kb.1 g teljes tejport vagy kb. 1,5 g félzsíros tejport, illetve sovány tejport közvetlen beméréssel vagy visszaméréssel 0,1 mg pontossággal az extraháló edénybe (5.2.) mérünk. 10 ml vizet adunk hozzá és a tejpor teljes feloldásáig rázogatjuk (egyes mintáknál melegítésre is szükség lehet).
6.2.3. 1,5 ml ammónia-oldatot (4.1.) vagy töményebb oldat ezzel egyenértékű térfogatát adjuk hozzá, vízfürdőn (5.6.) időnként összerázva 15 percig 60–70 °C-on melegítjük, majd folyóvízzel lehűtjük.
6.2.4. 10 ml etanolt (4.2.) adunk hozzá és a folyadékokat a nyitott extraháló edényben óvatosan összekeverjük.
6.2.5. 25 ml dietil-étert (4.3.) adunk hozzá és folyó vízzel lehűtjük. A készüléket lezárjuk, 1 percen keresztül erősen rázzuk és közben többször megfordítjuk.
6.2.6. A dugót óvatosan kivesszük és 25 ml petrolétert (4.4.) adunk hozzá úgy, hogy néhány ml-ével leöblítjük a dugót és az extraháló edény nyakának belső falát az öblítőfolyadékot az extraháló edénybe folyatjuk. Az edényt a dugóval újra lezárjuk, 30 másodpercig rázzuk és időnként megfordítjuk. Ha a 6.2.7. pontban leírt műveletben nem centrifugálunk, nem szabad túl erősen rázni.
6.2.7. A vizsgálati anyagot állni hagyjuk, a felső folyadék fázis kitisztulásáig és a vizes fázistól való teljes elválásáig. A fázisok elválasztását megfelelő centrifugával (5.8.) is elvégezhetjük.
Megjegyzés:
Az a centrifuga, amely nem háromfázisú motorral működik, szikrát kelthet. Ezért különösen ügyelni kell arra, hogy robbanás vagy tűz ne keletkezzék az étergőzök jelenléte miatt (például a lombik eltörése esetén).
6.2.8. A dugót kivesszük, néhány ml oldószer eleggyel (4.5.) a dugót és a lombik nyakának belső oldalát leöblítjük, az öblítőfolyadékot az exraháló edénybe folyatjuk. A felső réteget dekantálással vagy pipettával (5.7.) gondosan és lehetőleg teljes mennyiségében a lombikba visszük át (6.2.1.).
Megjegyzés:
Ha a leszivatást nem pipettával végezzük, szükség lehet arra, hogy valamennyi vizet hozzáadva a két folyadék közti fázishatárt, a dekantálás megkönnyítésére, megemeljük.
6.2.9. Az extraháló edény nyakának külső és belső falát, vagy a pipetta csúcsát és alsó részét néhány ml oldószer eleggyel leöblítjük. Az extraháló edény külső oldaláról az öblítőfolyadékot a lombikba, a pipetta és a belső fal öblítőfolyadékát pedig az extraháló edénybe folyatjuk.
6.2.10. A 6.2.5–6.2.9. műveletek megismétlésével 15 ml dietil-éter és 15 ml petroléter felhasználásával második extrakciót végzünk.
6.2.11. Harmadik extrakciót végzünk a 6.2.10. szerint, azonban az utolsó öblítést (6.2.9.) elhagyjuk.
Megjegyzés:
Sovány tejpor esetében harmadik extrakció nem szükséges.
6.2.12. Az oldószert (beleértve az etanolt is) gondos elpárologtatással vagy desztillációval eltávolítjuk. Ha a lombik űrtartalma kicsi, akkor minden extrakció után a fent leírt módon a kis mennyiségű oldószert eltávolítjuk.
6.2.13. Ha már nem érzünk oldószerszagot, a lombikot fektetve 1 órára a szárítószekrénybe helyezzük és szárítjük.
6.2.14. A lombikot a mérlegszoba hőmérsékletére hűtjük és 0,1 mg pontossággal mérjük.
6.2.15. A 6.2.13. és a 6.2.14. műveleteket addig ismételjük, míg a két egymás utáni mérés különbsége 0,5 mg-nál kisebb lesz vagy a tömeg növekszik. Tömegnövekedés esetén a számolásnál (7.) a legkisebb értéket vegyük figyelembe. A végső tömeget M1-gyel jelöljük.
6.2.16. Ezután 15–25 ml petrolétert adunk hozzá annak vizsgálatára, hogy az extrahált anyag teljesen oldható-e. Az oldószert enyhén melegítjük és rázogatjuk, amíg az egész zsír fel nem oldódik.
6.2.16.1. Ha az extrahált anyag petroléterben teljesen oldható, a 6.2.1. és 6.2.15. mérések közti különbség a zsír tömege.
6.2.16.2. Kétség esetén vagy ha oldhatatlan anyagok vannak jelen, a lombikban lévő zsírt meleg petroléteres mosással ismét extraháljuk, és az oldhatatlan részt minden dekantálás előtt ülepítjük. A lombik nyakának külső oldalát háromszor leöblítjük. A lombikot 1 órán keresztül fekvő helyzetben szárítószekrényben szárítjuk, 6.2.1. szerint a mérlegszoba hőmérsékletére hűtjük, majd 0,1 mg pontossággal mérjük. A zsír mennyisége a 6.2.15. szerinti mennyiség és ennek a tényleges végső mennyiségnek a különbsége.
7. Az eredmények kiszámítása
Az extrahált zsír grammban kifejezett tömegét a következő képlettel számítjuk ki:
(M1 – M2) – (B1 – B2)
A minta zsírtartalma %-ban:
ahol:
M1 = a lombik tömege zsírral együtt g-ban a 6.2.15 művelet után,
M2 = a lombik tömege g-ban a 6.2.1. művelet után, illetve oldhatatlan anyag jelenléte vagy annak gyanúja esetében a 6.2.16.2. művelet után,
B1 = a vakpróba lombikjának tömege g-ban a 6.2.15. művelet után,
B2 = a vakpróba lombikjának tömege g-ban a 6.2.1. művelet után, illetve oldhatatlan anyag jelenléte vagy annak gyanúja esetén a 6.2.16.2. művelet után,
S = a bemért minta mennyisége g-ban.
8. Ismételhetőség
A párhuzamos vizsgálatok eredményei között, azaz ugyanazon vizsgáló által, ugyanabból a mintából egyidejűleg vagy közvetlenül egymás után végzett vizsgálati eredmény között a megengedett eltérés legfeljebb 0,2 g zsír/100 g termék, sovány tejpor esetében 0,1 g zsír/100 g termék lehet.
5. módszer: Szacharóztartalom meghatározás (polarimetriás módszer)
1. Tárgy és alkalmazási terület
Ezzel a módszerrel határozzuk meg a szacharóztartalmat a következő termékekből:
– cukrozott sűrített tej,
– cukrozott sűrített félzsíros tej,
– cukrozott sűrített sovány tej,
– A minta nem tartalmazhat invertcukrot.
2. Fogalommeghatározás
A cukrozott sűrített tej szacharóztartalma: az ezzel a módszerrel meghatározott szacharóztartalom.
3. A módszer elve
A módszer a Clerget-inverzió elvén alapszik: gyengén savas kezelés hatására a szacharóz teljesen hidrolizál, míg a laktóz vagy más cukrok nem változnak. A szacharóztartalmat az oldat forgatóképességének változásából állapítjuk meg.
A minta tiszta szűrletét a laktóz okozta mutarotáció nélkül úgy állítjuk elő, hogy az oldatot ammóniával kezeljük, majd semlegesítjük, és cink-acetát, illetve kálium-hexaciano-ferrát/II/-oldat egymás után történő hozzáadásával csapadékot képzünk.
A szűrlet egy részében a szacharózt megfelelő eljárással hidrolizáljuk.
A szűrlet inverzió előtti és utáni forgatóképességéből számítjuk ki a szacharóztartalmat a megfelelő képlet alapján.
4. Vegyszerek
4.1. Cink-acetát oldat, 1 mólos: 21,9 g kristályos cink-acetátot (Zn(C2H3O2)2 . 2H2O) és 3 ml jégecetet vízzel 100 ml-re feltöltünk.
4.2. Kálium-hexaciano-ferrát/II/ oldat 0,25 mólos : 10,6 g kristályos kálium-hexaciano-ferrát/II/-ot (K4(Fe(CN)6) . 3H2O vízzel 100 ml-re feltöltünk.
4.3. Sósav-oldat, 6,35 + 0,20 mólos (20–22%), vagy 5,0 + 0,2 mólos (16–18%)
4.4. Ammónia-oldat, 2,0 + 0,2 mólos, kb. 3,5% (m/m)
4.5. Ecetsav-oldat, 2,0 + 0,2 mólos, 12% (m/m)
4.6. Brómtimolkék-indikátor 1%-os (m/v) etanolos oldata.
5. Eszközök
5.1. Mérleg, 10 mg-os pontosságú
5.2. Polarimétercső, 2 dm-es, pontosan kalibrált hosszúságú
5.3. Polariméter vagy szachariméter:
a) polariméter nátriumfénnyel vagy zöld higanyfénnyel (higanygőzlámpa prizmával vagy speciális Wrattenszűrővel Nr. 77A), legalább 0,05 szögfok leolvasási pontossággal,
b) szachariméter nemzetközi cukorskálával, 6%-os kálium-bikromát oldat 15 mm vastag szűrőjén átvezetett fehér fénnyel vagy nátriumfénnyel. A nemzetközi cukorskálán a leolvasási pontosság legalább 0,1 fok legyen.
5.4. Vízfürdő, 60±1 °C-ra beállítható.
6. A vizsgálat menete
6.1. Ellenőrző vizsgálat
A módszer, a reagensek és a készülékek ellenőrzésére a következő módszerrel kettős ellenőrző vizsgálatot végzünk 100 g tej, vagy 110 g soványtej és 18,00 g tiszta szacharóz felhasználásával. Ez a keverék 40,00 g 45% szacharóztartalmú sűrített tejnek felel meg. A cukortartalmat a 7. pontban megadott képlettel számítjuk ki, ahol az 1. képletben szereplő M, F és P a bemért tejmennyiséget, a tej zsír- és fehérjetartalmát jelenti, a 2. képletben szereplő M helyére 40,00 számot kell beírni. A megállapított értékek középértéke a tényleges 45%-tól legfeljebb 0,2%-kal térhet el.
6.2. Meghatározás
6.2.1. A jól összekevert mintából kb. 40 g-ot 10 mg pontossággal 100 ml térfogatú főzőpohárba mérünk. 50 ml forróvizet (80–90 °C) adunk hozzá és alaposan összekeverjük.
6.2.2. A keveréket 200 ml-es mérőlombikba maradéktalanul áttöltjük és a főzőpoharat kis mennyiségű desztillált vízzel többször átöblítjük. A desztillált víz hőmérséklete 60 °C, összes térfogata 120–150 ml legyen. Összekeverjük és szobahőfokra hűtjük.
6.2.3. 5 ml hígított ammónia-oldatot (4.4.) adunk hozzá, újra összekeverjük és 15 percig állni hagyjuk.
6.2.4. Az ammóniát ekvivalens mennyiségű ecetsav-oldat (4.5.) hozzáadásával semlegesítjük. Előzetesen a pontos mennyiséget az ammónia-oldat brómtimolkék indikátor (4.6.) jelenlétében történő titrálásával állapítjuk meg. Összekeverjük az oldatot.
6.2.5. 12,5 ml cink-acetát oldatot (4.1.) adunk hozzá, miközben a megdöntött lombikot körkörösen forgatjuk.
6.2.6. Az acetát-oldat adagolásával azonos módon 12,5 ml kálium-hexaciano-ferrát (4.2.) oldatot adunk hozzá.
6.2.7. A lombik tartalmát 20 °C-ra beállítjuk és 20 °C-os vízzel a 200 ml-es jelig töltjük.
Megjegyzés:
Eddig a műveletig a víz vagy a reagensek adagolását úgy kell végezni, hogy a légbuborékok képződését elkerüljük. Ugyanebből a célból minden keverést a lombik óvatos körkörös forgatásával és nem rázogatással kell végezni. A 200 ml végtérfogatra való feltöltést megelőzően keletkezett légbuborékokat a lombikra kapcsolt vákuumszivattyúval és a lombik körkörös mozgatásával távolítjuk el.
6.2.8. A lombikot száraz dugóval lezárjuk, és erős rázással alaposan összekeverjük.
6.2.9. Néhány percig állni hagyjuk, majd száraz szűrőpapíron átszűrjük, a szürlet első 25 ml-ét kiöntjük.
6.2.10. Közvetlen polarizáció: meghatározzuk a szürlet optikai forgatóképességét 20±1 °C-on.
6.2.11. Inverzió: az előbbiek szerint nyert szürletből 40 ml-t 50 ml-es mérőlombikba pipettázunk. 6,0 ml 6,35 mólos vagy 7,5 ml 5,0 mólos sósavat (4.3.) adunk hozzá.
A lombikot 15 percre 60 °C-os vízfürdőbe állítjuk úgy, hogy a lombik a nyakáig belemerüljön. Az első 5 percben körkörös mozgatással keverjük, ezalatt a lombik tartalmának el kell érnie a vízfürdő hőmérsékletét. 20 °C-ra lehűtjük és 20 °C-os vízzel a 50 ml-es jelig feltöltjük. Összekeverjük és ezen a hőmérsékleten 1 órán keresztül állni hagyjuk.
6.2.12. Inverzió utáni polarizáció
Az invertált oldat forgatóképességét 20±0,2 °C-on mérjük. (Ha a polarizálócsőben a folyadék hőmérséklete több mint 0,2 °C-kal eltér a 20 °C-tól, akkor a 7.2. pontban megadott hőmérsékletkorrekciót kell alkalmazni.)
7. Az eredmények kiszámítása
7.1. Számítás
A szacharóztartalom kiszámítását a következő képletek szerint végezzük:
1. v= |
M |
⋅ (1,08F+1,55P) |
100 |
2. S= |
D–1,25I |
⋅ |
V–v |
⋅ |
V |
% |
Q |
V |
L ⋅ M |
ahol:
S = szacharóztartalom,
M = a bemért minta tömege g-ban,
F = a minta zsírtartalma %-ban,
P = a minta fehérjetartalma (N×6,38) %-ban,
V = az a térfogat ml-ben, amire a mintát a szűrés előtt felhígítjuk,
v = a szűréskor képződött csapadék térfogat korrekciója ml-ben,
D = a közvetlen polarizációs érték (az inverzió előtti polarizáció),
I = az inverzió utáni polarizációs érték,
L = a polarimétercső hossza dm-ben,
Q = inverziós faktor, amelynek az értékére a következőkben térünk ki.
Megjegyzések:
a) Ha pontosan 40,00 g sűrített tejet mértünk be, és nátriumlámpás polariméter szögfokot és 2 dm-es polarimétercsövet használunk 20±0,1 °C-on, akkor a normál sűrített tej (C=9) szacharóztartalmát a következő képlettel számítjuk ki:
S = (D–1,25 I) × (2,833–0,00612 F–0,00878 P)
b) Ha a polarizációt inverzió után a megadott 20 °C-tól (T) eltérő hőmérsékleten mértük, az értéket meg kell szorozni a következő faktorral: [1+0,0037 (T–20)]
7.2. Az inverziós faktor (Q) értéke
A következő képletek megadják a Q pontos értékét különböző fényforrások, különböző koncentrációk és hőmérsékletek esetén:
nátriumfénynél és polariméter szögfokban mérve:
Q = 0,8825+0,0006 (C–9)–0,0033 (T–20),
zöld higanyfénynél és polariméter szögfokban mérve:
Q = 1,0392+0,0007 (C–9)–0,0039 (T–20),
fehér fénynél bikromátszűrővel és nemzetközi cukorskálával ellátott szachariméterrel mérve:
Q = 2,549+0,0017 (C–9)–0,0095 (T–20).
Az előbbi képletekben a betűk jelentése:
C = a polarimetriás mérésnek megfelelő összes cukortartalom az invertált oldatban, százalékban,
T = az invertált oldat hőmérséklete a polarimetriás mérés alatt.
1. megjegyzés:
Az invertált oldatban lévő összes cukortartalmat %-ban (C) a közvetlen polarizációs érték és az inverzió utáni polarizációs érték a szacharóz, laktóz és invertcukor specifikus forgatóképességének normál értékével a szokásos módon lehet kiszámítani.
A 0,0006 (C–9) korrekció csak akkor pontos, ha C kb. = 9, szokásos sűrített tejnél ez a korrekció elhagyható, mivel C 9-hez közel van.
2. megjegyzés:
A 20 °C mérési hőmérséklethez képest 1 °C hőmérséklet eltérés a közvetlen leolvasásban keveset jelent, de az inverzió utáni leolvasásnál a 0,2 °C-nál nagyobb eltérés esetén már korrekcióra van szükség. A 0,0033 (T–20) korrekció stb. csak 18 °C–22 °C között pontos.
8. Ismételhetőség
A párhuzamos vizsgálatok eredményei között, azaz ugyanazon vizsgáló által, ugyanabból a mintából egyidejűleg vagy közvetlenül egymás után végzett vizsgálatok eredménye között a megengedett eltérés legfeljebb 0,3 g szacharóz/100 g termék lehet.
6. módszer: Tejsav és laktáttartalom meghatározás
1. Tárgy és alkalmazási terület
Ezzel a módszerrel a következő termékek tejsav- és laktáttartalmát határozzuk meg:
– emelt zsírtartalmú tejpor,
– teljes tejpor,
– félzsíros tejpor,
– sovány tejpor.
2. Fogalommeghatározás
A tejpor tejsav- és laktáttartalma: az ezzel a módszerrel meghatározott tejsav- és laktáttartalom, tejsavban kifejezve.
3. A módszer elve
Réz-szulfát és kalcium-hidroxid egyidejű adagolása után a zsírt, a fehérjét és laktózt szűréssel eltávolítjuk. A szürletben lévő tejsavat tömény kénsavval réz-szulfát jelenlétében acetaldehiddé alakítjuk. Az acetaldehidet 4-hidroxi-difenillel fotometriásan meghatározzuk. A tejsav és laktáttartalmat tejsav mg/100 g zsírmentes szárazanyag értékben adjuk meg.
4. Vegyszerek
4.1. Réz/II/szulfát oldat: 250 g réz/II/szulfátot (CuSO4 . 5H2O) vízben oldunk, és 1000 ml-re feltöltjük.
4.2. Kálcium-hidroxid-szuszpenzió:
4.2.1. 300 g kálcium-hidroxidot [Ca(OH)2] mozsárban összesen 900 ml víz felhasználásával eldörzsöljük. A szuszpenziót a felhasználás előtt frissen készítjük.
4.2.2. Másik változat: 300 g kálcium-hidroxidot [Ca(OH)2] mozsárban összesen 1400 ml víz felhasználásával eldörzsölünk. A szuszpenziót a felhasználás előtt frissen készítjük.
4.3. Kénsavas réz/II/szulfát oldat: 300 ml kénsavhoz (95,5–97,0% (m/m) H2SO4) 0,5 ml réz/II/szulfát oldatot (4.1.) adunk.
4.4. 4-hidroxi-difenil oldat (C6H5C6H4OH): 0,75 g 4-hidroxi-difenilt enyhe melegítés és keverés közben 5 gramm NaOH/100 ml koncentrációjú vizes nátrium-hidroxid oldat 5 ml-ében feloldunk. Mérőlombikban vízzel 50 ml-re feltöltjük. Az oldatot sötét, hűvös helyen, barna folyadéküvegben tároljuk. Ne használjuk, ha színváltozás vagy zavarosodás lép fel. Legfeljebb 72 óráig tárolható.
4.5. Tejsav kalibráló oldat: közvetlenül a felhasználás előtt 0,1067 g lítium-laktátot (CH3CHOHCOOLi) vízben feloldunk és mérőlombikban 1000 ml-re feltöltünk. Ennek az oldatnak 1 ml-e 0,1 mg tejsavnak felel meg.
4.6. Visszaállított normál tej: jó minőségű tejpor mintákat előzőleg megvizsgálunk. A kalibrációs görbe felvételéhez azokat a mintákat választjuk ki, amelyek tejsav tartalma a legkisebb, legfeljebb 30 mg/100 g zsírmentes szárazanyag tartalommal. A műveleteket a 6.2.1. és a 6.2.2. szerint végezzük.
5. Eszközök
5.1. Analitikai mérleg
5.2. Spektrofotométer, 570 nm hullámhosszra beállítható
5.3. Vízfürdő, 30+2 °C-ra beállítható
5.4. Dörzsmozsár törővel
5.5. Szürőpapír, (Schleicher és Schüll 595, Whatman 1 vagy azzal egyenértékű)
5.6. Reagensüveg Pyrex vagy azzal egyenértékű üvegből (mérete: 25x150 mm)
Megjegyzés:
Minden üvegedény teljesen tiszta legyen, és csak ehhez a vizsgálathoz használjuk. A csapadékot tartalmazó üvegedényeket tömény sósavas kiöblítés után mosogassuk el.
6. A vizsgálat menete
6.1. Vakpróba
Vakpróbát készítünk, amelyhez 30 ml vizet töltünk egy 50 ml-es mérőlombikba és a 6.2.4–6.2.11. műveleteket elvégezzük. Ha a vakpróba eredménye vízzel szemben mérve a 20 mg tejsav/100 mg zsírmentes szárazanyagnak megfelelő értéket túllépi, a reagenseket felül kell vizsgálni, a szennyezett reagenseket, illetve reagenst mással kell pótolni. A vakpróbát a minta vizsgálatával egyidejűleg kell végezni.
6.2. Meghatározás
Megjegyzés:
Különösen nyállal és izzadsággal való szennyeződéstől óvjuk a mintákat.
6.2.1. A minta zsírmentes szárazanyagtartalmát (a) megkapjuk, ha 100-ból levonjuk a zsírtartalmat (meghatározása a 4. módszerrel) és a víztartalmat (meghatározása a 2. módszerrel).
6.2.2. A minta |
1000 |
g-ját 0,1 g pontossággal lemérjük. |
a–10 |
Ehhez a mintamennyiséghez 100 ml vizet adunk és alaposan összekeverjük.
6.2.3. Az így kapott oldatból 5 ml-t 50 ml-es mérőlombikba pipettázunk és vízzel kb. 30 ml-re felhígítjuk.
6.2.4. Forgatás közben lassan hozzáadunk 5 ml réz-szulfát oldatot (4.1.) és 10 percig állni hagyjuk.
6.2.5. Forgatás közben lassan hozzáadunk 5 ml kálcium-hidroxid szuszpenziót (4.2.1.) vagy 10 ml kalcium-hidroxid szuszpenziót (4.2.2.).
6.2.6. Vízzel 50 ml-re feltöltjük, erősen összerázzuk, 10 percig állni hagyjuk, majd szűrjük. A szürlet első részét elöntjük. 6.2.7. A szürlet 1 ml-ét reagensüvegbe pipettázzuk (5.6.).
6.2.8. Bürettával vagy osztott pipettával a kénsav-réz-szulfát-oldatból (4.3.) 6,0 ml-t a reagensüvegbe adagolunk. Összekeverjük.
6.2.9. A reagensüveget 5 percen keresztül forró vízfürdőn melegítjük. Folyó vízzel szobahőfokra hűtjük.
6.2.10. Két csepp 4-hidroxi-difenil-reagenst (4.4.) adunk hozzá és erősen összerázzuk, hogy a reagens az egész folyadékban egyenletesen eloszoljon. A reagensüveget 30±2 °C hőmérsékletű vízfürdőbe helyezzük, 15 percen keresztül benne hagyjuk és időnként rázogatjuk.
6.2.11. A reagensüveget 90 másodpercre forró vízfürdőbe helyezzük, majd folyó vízzel szobahőmérsékletre hűtjük.
6.2.12. Az optikai sűrűséget a vakpróbával szemben (6.1.) 3 órán belül az 5.2. pontban megadott hullámhosszon mérjük.
6.2.13. Amennyiben az optikai sűrűség a kalibrációs görbe legmagasabb pontját túllépi, a 6.2.6. szerint nyert szürlet megfelelő hígításával kapott mintával a vizsgálatot megismételjük.
6.3. A kalibrációs görbe felvétele
6.3.1. A visszaállított tej (4.6.) 5 ml-ét 5 db 50 ml-es mérőlombikba pipettázzuk. Ezekbe a lombikokba 0, 1, 2, 3, 4 ml kalibráló oldatot (4.5.) pipettázunk. Az így kapott oldatok 0, 20, 40, 60, 80 mg tejsav/100 g zsírmentes szárazanyag koncentrációnak felelnek meg.
6.3.2. Vízzel kb. 30 ml-re hígítjuk és a 6.2.4–6.2.11. pontban leírtak szerint járunk el.
6.3.3. A standard (6.3.1.) optikai sűrűségét a vakpróbával (6.1.) szemben az 5.2. pontban megadott hullámhosszon mérjük. Az optikai sűrűség adatait a 6.3.1. pontban megadott 0 mg, 20 mg, 40 mg, 60 mg, 80 mg/100 g zsírmentes szárazanyagra vonatkoztatott tejsav mennyiségének függvényében diagramon ábrázoljuk. Ezeknek a pontoknak legjobban megfelelő egyenest megrajzoljuk, és a kalibrációs görbét úgy készítjük el, hogy az egyenes önmagával párhuzamosan eltolva a koordinátarendszer nullpontját metssze.
7. Az eredmények kiszámítása
A minta 6.2.12. vagy 6.2.13. szerint mért optikai sűrűségét a kalibrációs görbe segítségével tejsav mg/100 g zsírmentes szárazanyag értékre számoljuk át. Az eredményt a hígítási faktorral megszorozzuk, amennyiben a szürletet a 6.2.13. szerint hígítottuk.
8. Ismételhetőség
A párhuzamos vizsgálatok eredményei között, azaz ugyanazon vizsgáló által, ugyanabból a mintából egyidejűleg vagy közvetlenül egymás után végzett vizsgálati eredmények között a megengedett eltérés legfeljebb 8 mg tejsav/100 g zsírmentes szárazanyag lehet 80 mg/100 g mért értékig. Nagyobb értékeknél ez a különbség a legkisebb érték 10%-nál nem lehet nagyobb.
7. módszer: Foszfatáz-aktivitás meghatározás (módosított Sanders-Sagar-eljárás)
1. Tárgy és alkalmazási terület
Ez a módszer a foszfatáz-aktivitás meghatározását írja le a következő termékekben:
– emelt zsírtartalmú tejpor,
– teljes tejpor,
– félzsíros tejpor,
– sovány tejpor.
2. Fogalommeghatározás
A foszfatáz-aktivitás a jelenlévő aktív, alkalikus foszfatáz mennyiségének a mértéke. Ezt az értéket az 1 ml hígított tejből, az itt leírt módszerrel felszabadított fenol µg-ban állapítjuk meg.
3. A módszer elve
A tejpor foszfatáz-aktivitását a foszfatáz azon képessége alapján határozzuk meg, hogy dinátrium-fenil-foszfátból fenolt képes felszabadítani. Az alább leírt feltételek közt felszabaduló fenol mennyiségét spektrofotometriás méréssel, a Gibb reagenssel kialakított szín mérése útján határozzuk meg.
4. Vegyszerek
4.1. A oldat
Bárium-borát-hidroxid-puffer: pH 10,6±0,1 20 °C-on. 25,0 g bárium-hidroxidot (Ba(OH)2 . 8H2O) vízben oldunk és 500 ml-re felhígítjuk. 11,0 g bórsavat (H3BO3) vízben feloldunk és 500 ml-re felhígítjuk.
Mindkét oldatot 50 °C-ra felmelegítjük és összekeverjük. Az elegyet rázogatva szobahőmérsékletre hűtjük. A pH-értékét bárium-hidroxid-oldattal 10,6±0,1-re állítjuk be és leszűrjük. Az oldatot jól zárható edényben tároljuk. A puffert felhasználás előtt azonos mennyiségű vízzel felhígítjuk.
4.2. B oldat
Színképző puffer: 6,0 g nátrium-metaborátot (NaBO2) vagy (12,5 g NaBO2 . 4H2O) és 20 g nátrium-kloridot (NaCl) vízben feloldunk és 1000 ml-re feltöltjük.
4.3. C oldat
Szubsztrátum-puffer oldat
4.3.1. 0,5 g dinátrium-fenil-foszfátot (Na2C6H5PO4 . 2H2O) 4,5 ml B oldatban (4.2.) feloldunk. Az E oldatból (4.5.) 2 cseppet adunk hozzá, és 30 percig állni hagyjuk. A színanyagot 2,5 ml butanollal (4.10.) extraháljuk. Amennyiben szükséges, a színanyag extrakciót megismételjük. A kivonás után a butanolt kiöntjük. Az oldat hűtőszekrényben több napig eltartható. A felhasználás előtt a színezéket újra kifejlesztjük és extraháljuk.
4.3.2. Ebből az oldatból 1 ml-t egy 100 ml-es mérőlombikba viszünk át és az A oldattal jelig töltjük. A pufferoldatot közvetlenül a felhasználás előtt készítjük el.
4.4. D oldat
Csapadékképző reagens: 3,0 g cink-szulfátot (ZnSO4 . 7H2O) és 0,6 g réz-szulfátot (CuSO4 . 5H2O) vízben feloldunk, és 100 ml-re feltöltjük.
4.5. E oldat
Gibb reagens: 0,040 g 2,6-dibrómkinon-1,4-klórimidet (C6H2Br2ClNO) 10 ml 96%-os etil-alkoholban feloldunk. Az oldatot sötét üvegben hűtőszekrényben tároljuk. Elszíneződött reagens nem használható.
4.6. Színhígító puffer
A színképző B oldatból (4.2.) 10 ml-t vízzel 100 ml-re hígítunk.
4.7. Réz-szulfát oldat
0,05 g réz(II)-szulfátot (CuSO4 . 5H2O) vízben oldunk és vízzel 100 ml-re feltöltjük.
4.8. Fenol törzsoldat
0,200±0,001 g tiszta fenolt vízben oldunk és mérőlombikban 100 ml-re feltöltjük. Az oldat hűtőszekrényben néhány hónapig eltartható. Ebből az oldatból 10 ml-t vízzel 100 ml-re feltöltünk. Ezen hígított oldat 1 ml-e 200 µg fenolt tartalmaz és további hígítások előállításához is felhasználható.
4.9. Kiforralt desztillált víz
4.10. n-butanol
5. Eszközök
5.1. Analitikai mérleg
5.2. Vízfürdő, 37±1 °C-ra beállítható
5.3. Spektrofotométer, 610 nm hullámhossznál történő méréshez
5.4. Szűrőpapír (Schleicher és Schüll 597, Whatman 42 vagy más, azonos minőségű szűrőpapír)
5.5. Vízfürdő, forrásban lévő
5.6. Alumíniumfólia
6. A minta előkészítése
A minta 10 g-ját (0,1 g pontossággal lemérve) 90 ml vízben feloldjuk. A por feloldása közben a hőmérséklet semmiképpen sem lehet 35 °C-nál nagyobb.
7. A vizsgálat menete
7.1. Óvórendszabályok
1. Kerüljük a napfény hatását.
2. Minden üvegeszközt, a dugókat és a felszereléseket állandóan tisztán kell tartani, vízzel leöblíteni és kifőzni, vagy gőzzel kezelni.
3. A műanyagok alkalmazását (például dugók) kerüljük, mivel azok fenolt tartalmazhatnak.
4. A nyál foszfatázt tartalmaz; a nyálnyomokkal való szennyeződést gondosan kerülni kell.
7.2. Meghatározás
7.2.1. Két reagensüvegbe a 6. szerint előkészített, visszaállított tejből 1-1 ml-t adagolunk.
7.2.2. A reagensüvegek egyikét 2 percen keresztül forrásban lévő vízfürdőben melegítjük. A reagensüveget és a vízfürdőt (5.5.) egy alufóliával bevont főzőpohárral (5.6.) lefedjük, annak biztosítására, hogy a reagensüveg teljes egészében átforrósodjon. Hideg vízzel szobahőfokra hűtjük. Ezt a reagensüveget a vakpróbához használjuk. Ettől a ponttól kezdődően mindkét reagensüveget azonos módon kezeljük.
7.2.3. 10 ml C oldatot (4.3.2.) adunk hozzá, összekeverjük, és a reagensüvegeket 37 °C-os vízfürdőbe (5.2.) állítjuk.
7.2.4. 60 percig vízfürdőben inkubáljuk és időnként összerázzuk.
7.2.5. A reagensüvegeket rögtön a forrásban lévő vízfürdőbe (5.5.) állítjuk és 2 percig hevítjük, hideg vízzel szobahőmérsékletre hűtjük.
7.2.6. A D oldatból 1 ml-t (4.4.) hozzáadunk, összekeverjük, és száraz szűrőpapíron szűrjük, az első szürleteket mindaddig elöntjük, amíg tiszta folyadékot nem kapunk.
7.2.7. Mindkét szürletből 5 ml-t reagensüvegekbe adagolunk, 5 ml B oldatot (4.2.) és 0,1 ml E oldatot (4.5.) adunk hozzá. Összekeverjük.
7.2.8. A szín kialakulásához 30 percig szobahőfokon napfénytől védve állni hagyjuk.
7.2.9. Az optikai sűrűséget a vakpróbával szemben az 5.3. alatt megadott hullámhosszon mérjük.
7.2.10. A meghatározást megismételjük, ha az oldat optikai sűrűsége az 5.3. pont szerint kapott 20 mg-os fenol standardénál nagyobb. Ha ezt az értéket túlléptük, a visszaállított tej (6.) térfogatát olyan tej megfelelő térfogatával hígítjuk fel, amelyet a 7.2.2. szerint a foszfatáz inaktiválására gondosan felforraltunk.
7.3. A kalibrációs görbe felvétele
7.3.1. 4 db 100 ml-es mérőlombikba 1, 3, 5 és 10 ml-t mérünk be a 4.8. szerint felhígított kalibráló oldatból és vízzel jelig töltjük. Ezek a hígítások ml-enként 2, 6, 10 és 20 µg fenolt tartalmaznak.
7.3.2. 1 ml vizet, illetve a referenciaoldatok 1 ml-ét (7.3.1.) a reagensüvegekbe pipettázzuk, hogy olyan mintasorozatot kapjunk, amely 0, 2, 6, 10 és 20 µg fenolt tartalmaz.
7.3.3. Egymás után minden reagensüvegbe 1 ml réz-szulfát-oldatot (4.7.), 5 ml színhigító puffert (4.6.), 3 ml vizet és 0,1 ml E oldatot (4.5.) adunk. Összekeverjük.
7.3.4. A reagensüvegeket szobahőmérsékleten napfénytől védve 30 percig állni hagyjuk.
7.3.5. A reagensüvegekből kivett oldatok optikai sűrűségét a vakpróbahoz viszonyítva az 5.3. pontban megadott hullámhosszon megmérjük.
7.3.6. Az optikai sűrűséget a 7.2. pontban megadottak szerint µg-ban mért fenol mennyiség függvényében kalibrációs görbén ábrázoljuk.
8. Az eredmények kiszámítása
8.1. A 7.2.9. szerint kapott értékeket a kalibrációs görbe felhasználásával µg fenolra számítjuk át.
8.2. A µg fenolban kifejezett foszfatáz-aktívitást a visszaállított tej 1 ml-ére vonatkoztatva a következő képlettel számítjuk ki:
Foszfatáz-aktivitás = 2,4×P
ahol
P = a fenolmennyiség µg-ban a 8.1. szerint.
8.3. Ha a 7.2.10. szerinti hígítás szükséges volt, úgy a 8.2. szerinti eredményeket még a hígítási faktorral is meg kell szorozni.
9. Ismételhetőség
Párhuzamos vizsgálatok eredményei között, azaz ugyanazon vizsgáló által, ugyanazon mintából egyidejűleg vagy közvetlenül egymás után végzett vizsgálatok eredménye közt a megengedett eltérés legfeljebb 2 µg felszabadított fenol/ml visszaállított tej lehet.
8. módszer: Foszfatáz-aktivitás meghatározása (Aschaffenburg–Mullen-módszer)
1. Tárgy és alkalmazási terület
Ez a módszer a foszfatáz-aktivitás meghatározását írja le a következő termékekben:
– zsíros tejpor,
– teljes tejpor,
– félzsíros tejpor,
– sovány tejpor.
2. Fogalommeghatározás
A tejpor foszfatáz-aktivitása a termékben lévő aktív alkalikus foszfatáz mennyiségének a mértéke. Ezt az értéket a visszaállított minta 1 ml-éből az itt leírt módszerrel felszabaduló p-nitrofenol µg-ban határozzuk meg.
3. A módszer elve
A visszaállított mintát 10,2 pH-jú szubsztrát-pufferrel felhígítjuk és 37 °C hőmérsékleten termosztáljuk. A mintában lévő alkalikus foszfatáz ezen körülmények között a dinátrium-p-nitrofenil-foszfátból p-nitrofenolt szabadít fel. A felszabadított p-nitrofenolt standard színes üvegekkel való közvetlen összehasonlítással, egyszerű komparátorban, visszavert fényben meghatározzuk.
4. Vegyszerek
4.1. Nátrium-karbonát-hidrogénkarbonát puffer
3,5 g vízmentes nátrium-karbonátot és 1,5 g nátrium-hidrogénkarbonátot vízben feloldunk és mérőlombikban 1000 ml-re feltöltjük.
4.2. Szubsztrát-puffer-oldat
1,5 g dinátrium-p-nitrofenil-foszfátot nátrium-karbonát/hidrogénkarbonát pufferben (4.1.) feloldunk, és mérőlombikban a pufferrel (4.1.) 1000 ml-re feltöltjük. Az oldat egy hónapig stabilis marad, ha hűtőszekrényben (4 °C-on vagy az alatti hőfokon) tároljuk, azonban a tárolt oldatból színvizsgálatot kell végezni, lásd: 7. pont 3. megjegyzés.
4.3. Csapadékképző reagensek
4.3.1. Cink-szulfát oldat
30,8 g cink-szulfátot vízben feloldunk és mérőlombikban 100 ml-re feltöltjük.
4.3.2. Kálium-ferrocianid oldat
17,2 g kálium-ferrocianid-trihidrátot vízben feloldunk és mérőlombikban 100 ml-re feltöltjük.
5. Eszközök
5.1. Analitikai mérleg
5.2. Vízfürdő, termosztáttal 37±1 °C-ra beállítható
5.3. Komparátor, különleges p-nitrofenol µg/ml tej értékre hitelesített, standard, színes üveg tárcsával és 2 db, 25 mm-es küvettával.
6. A minta előkészítése
10 g tejport 90 ml vízben feloldunk. A tejpor oldási hőmérséklete nem lépheti túl a 35 °C-t.
7. A vizsgálat menete
Óvintézkedések:
1. Felhasználás után a reagensüvegeket ki kell üríteni, vízzel kiöblíteni, lúgos mosószert tartalmazó forró vízben kimosni, és tiszta forró vízzel kiöblíteni. Felhasználás előtt vízzel ki kell öblíteni és megszárítani. Használat után a pipettákat alaposan, tiszta hideg folyóvízben rögtön ki kell öblíteni, végül a felhasználás előtt vízzel ki kell öblíteni és megszárítani.
2. A reagensüvegeket felhasználás után rögtön forró folyóvízzel alaposan le kell öblíteni, majd 2 percen keresztül vízben kifőzni.
3. A szubsztrát-puffer-oldat (4.2.) legalább 1 hónapig stabil marad, ha azt hűtőszekrényben 4 °C-on vagy annál kisebb hőmérsékleten tároljuk. Az esetleges instabilitást sárga szín megjelenése jelzi. Vizsgálat közben a minták mellett mindig azonos mennyiségű szubsztrat-puffer oldatot tartalmazó kontroll mintát is olvassunk le. Az oldatot ne használjuk, ha olyan elszíneződés lép fel, amely a komparátor 25 mm-es küvettájában desztillált vízzel szemben mérve 10 µg-nál nagyobb értéknek felel meg.
4. Minden mintához külön pipettát használjunk, hogy a pipetta nyállal való szennyeződését elkerüljük.
5. A mintát a legrövidebb időre sem szabad közvetlen napfény hatásának kitenni.
7.1. Meghatározás
7.1.1. A szubsztrát-puffer 15 ml-ét tiszta, száraz reagens üvegbe pipettázzuk, majd hozzáadjuk a vizsgálandó visszaállított minta (6.) 2 ml-ét. A reagensüveget lezárjuk, forgatással összekeverjük, és 37 °C hőmérsékletű vízfürdőbe tesszük.
7.1.2. Egyidejűleg egy másik reagensüvegben lévő 15 ml szubsztrát-pufferhoz 2 ml kiforralt, visszaállított mintát adunk és a vizsgálandó mintával azonos módon vízfürdőbe helyezzük.
7.1.3. 2 óra múlva mindkét reagensüveget a vízfürdőből kivesszük, 0,5 ml cink-szulfát oldatot (4.3.1.) adunk hozzá, a dugóval az üvegeket lezárjuk, erősen összerázzuk és 3 percig állni hagyjuk. Hozzáadunk 0,5 ml kálium-ferrocianid oldatot (4.3.2.), alaposan összekeverjük, szűrjük, a tiszta szürletet tiszta reagensüvegben felfogjuk.
7.1.4. A szürletet 25 mm-es küvettába visszük, és a kifőzött minta szürletével a komparátorban speciális tárcsa (5.3.) segítségével összehasonlítjuk.
8. A eredmények kiszámítása
A 7.1.4. szerint közvetlenül leolvasott értékeket µg p-nitrofenol/ml visszaállított mintára adjuk meg.
9. Ismételhetőség
Párhuzamos vizsgálatok között, azaz ugyanazon vizsgáló által, ugyanazon mintából egyidejűleg vagy közvetlenül egymás után végzett két vizsgálat eredménye között a megengedett eltérés legfeljebb 2 µg nitrofenol/ml visszaállított tej lehet.
MAGYAR ÉLELMISZERKÖNYV
(Codex Alimentarius Hungaricus)
3–1–85/503 számú előírás
(2. kiadás – 2006.)
Étkezési kazeinek és kazeinátok vizsgálati módszerei
Ez az előírás 2007. július 1-jén lép hatályba, ezzel egyidejűleg a Magyar Élelmiszerkönyvnek az étkezési kazeinek és kazeinátok vizsgálatáról szóló 3–1–85/503 számú előírása 1995-ben jóváhagyott 1. kiadása hatályát veszti.
Ez az előírás az étkezési kazeinek és kazeinátok vizsgálati módszereiről szóló, 1985. október 25-i 85/503/EGK bizottsági irányelvnek való megfelelést szolgálja.
1. számú melléklet a 3–1–85/503 számú előírás 2. kiadásához
Étkezési kazeinek és kazeinátok analitikai vizsgálata
I. Minta-előkészítés és általános követelmények
II. Víztartalom meghatározás
III. Fehérjetartalom meghatározás
IV. Titrálható savasság meghatározás
V. Hamutartalom meghatározás (P2O5-ot beleértve)
VI. pH-érték meghatározás
2. számú melléklet a 3–1–85/503 számú előírás 2. kiadásához
Analitikai módszerek az étkezési kazein és kazeinát összetételének meghatározásához
Minta-előkészítés és általános követelmények
1. Minta előkészítése
1.1. Általános követelmények
A laboratóriumi vizsgálatokhoz rendelkezésre álló minta mennyisége legalább 200 g legyen.
1.2. A minta előkészítése a laboratóriumi vizsgálathoz
1.2.1. A mintát az edény állandó rázásával és forgatásával gondosan összekeverjük és az összes csomót eloszlatjuk (ehhez a mintát légmentesen zárható és elegendő befogadóképességű, a mintánál kétszer nagyobb térfogatú edénybe tesszük).
1.2.2. Az alaposan összekevert mintából (1.2.1.) 50 g körüli reprezentatív mennyiséget a szitába (3.3.) teszünk.
1.2.3. Ha az 50 g-ot a szita (3.3.) teljesen vagy majdnem teljesen [legalább 95% (m/m)-át] átengedi, úgy az 1.2.1. szerint járunk el.
1.2.4. Ellenkező esetben az 50 g-ot egy darálóval (3.4.) addig aprítjuk, amíg a szitálási előírásoknak meg nem felel (1.2.3.). Az átszitált minta teljes mennyiségét rögtön légmentesen zárható és megfelelő méretű edénybe öntjük (a méret a minta térfogatának kétszerese legyen) és állandó rázással és forgatással összekeverjük.
Ügyeljünk arra, hogy a folyamat alatt a minta nedvességtartalma ne változzon.
1.2.5. Ha a mintát előkészítettük, a vizsgálatot a lehető leggyorsabban el kell végezni.
1.3. Tárolóedény
A mintát lég- és nedvességmentesen zárható edényben kell tárolni.
2. Vegyszerek
2.1. Vízre vonatkozó előírás
2.1.1. Oldás, hígítás vagy mosás esetén mindig desztillált vizet vagy azzal azonos minőségű vizet használjunk.
2.1.2. Ahol az „oldás” vagy „hígítás” szó szerepel minden magyarázat nélkül, ott mindig „vizes oldatot” vagy „vízzel való hígítást” értünk.
2.2. Vegyszerek tisztasága
Minden vegyszer analitikailag legtisztább minőségű legyen, ha nincs más előírás.
3. Eszközök
3.1. A szokásos laboratóriumi eszközök, különösképpen:
3.2. Analitikai mérleg, legalább 0,1 mg leolvasási pontossággal
3.3. Ellenőrző szita
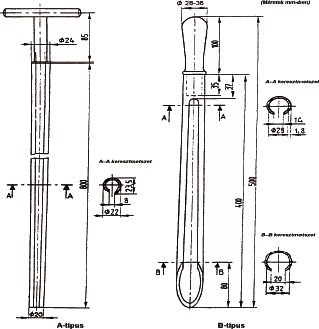
Fedővel lezárt, fémhuzalszövet szita, átmérője 200 mm, névleges lyukbősége 500 µm. A szitára és a huzalátmérőre megengedett eltérést az ISO 3310/1 szabvány tartalmazza. (Ellenőrző sziták. Műszaki követelmények és vizsgálat I. rész: Fémhuzalszövet ISO 3310/l–1975). A szitához egy gyűjtőedény tartozik.
3.4. Daráló, amivel szükség esetén (1.2.4.) a laboratóriumi minta túlzott felmelegedés, nedvességcsökkenés vagy -növekedés nélkül felaprítható.
Kalapácsos daráló nem alkalmazható.
4. Az eredmények megadása
A vizsgálati jegyzőkönyvben megadott eredmény legalább két olyan meghatározás középértéke legyen, amelyek ismételhetősége az adott analitikai módszernek megfelel.
Ha nincs más előírás, az eredményeket tömegszázalékban [% (m/m)] kell megadni.
5. Vizsgálati jegyzőkönyv
A vizsgálati jegyzőkönyvben az analitikai módszert és az eredményeket kell megadni. Kiegészítésképpen meg kell adni minden olyan részletet, amelyet a módszer nem ír elő vagy választható, valamint azokat a körülményeket, amelyek az eredményeket befolyásolhatták. A jegyzőkönyvnek a minta azonosításához szükséges minden adatot tartalmaznia kell.
1. módszer: A víztartalom meghatározás
1. Tárgy és alkalmazási terület
Ez a módszer a
– savas kazeinek,
– oltós kazeinek,
– kazeinátok
víztartalmának meghatározására szolgál.
2. Fogalommeghatározás
A kazein és kazeinát víztartalma: a leírt módszer szerinti tömegveszteség.
3. A módszer elve
102±1 °C hőmérsékletű szárítószekrényben légköri nyomáson tömegállandóságig szárítva a minta visszamaradt tömegét határozzuk meg. A tömegveszteséget a minta tömegének százalékában adjuk meg.
4. Eszközök
4.1. Analitikai mérleg, 0,1 mg pontosságú
4.2. Bemérőedények lapos aljjal, a kísérleti körülmények között nem rozsdásodó anyagból, pl. nikkelből, alumíniumból, saválló acélból vagy üvegből. Fedelük szorosan záró, de könnyen levehető legyen. Megfelelő méretek: 60–80 mm-ig, magasság kb. 25 mm.
4.3. Szárítószekrény, légköri nyomáson működő, jól szellőző, 102±1 °C hőmérsékletre termosztáttal beállítható. A hőmérséklet a szekrény minden pontján azonos legyen.
4.4. Exszikkátor frissen aktívált nedvességindikátort tartalmazó szilikagéllel, vagy azonos hatékonyságú szárítóközeggel.
4.5. Megfelelő szerszám az edények mozgatásához, pl. laboratóriumi edényfogó.
5. A vizsgálat menete
5.1. A vizsgálandó minta előkészítése
Lásd: I. Mintaelőkészítés és általános követelmények 1.2. pont.
5.2. A bemérőedény előkészítése
5.2.1. A fedetlen bemérőedényt és fedelét (4.2.) a 102±1 °C hőmérsékletre beállított szárítószekrényben (4.3.) legalább 1 óra hosszat szárítjuk.
5.2.2. Ezután a fedelet az edényre helyezzük, a lezárt edényt exszikkátorba (4.4.) tesszük és a mérlegszoba hőmérsékletére hűtjük, majd 0,1 mg pontossággal mérjük (M0).
5.3. Bemérés
3–5 g vizsgálandó mintát (5.1.) a bemérőedénybe mérünk, az edényt a fedéllel lefedjük és tartalmát 0,1 mg pontossággal lemérjük (M1).
5.4. Meghatározás
5.4.1. A fedetlen bemérőedényt és a fedelet 102±1 °C hőmérsékletre beállított szárítószekrénybe (4.3.) helyezzük 4 órára.
5.4.2. A fedelet újra az edényre helyezzük, az exszikkátorba (4.4.) tesszük, a mérlegszoba hőmérsékletére hűtjük, majd 0,1 mg pontossággal mérjük.
5.4.3. Végül a fedetlen edényt fedéllel együtt a szárítószekrényben 1 óra hosszat melegítjük, majd az 5.4.2. szerinti műveleteket megismételjük.
5.4.4. Ha az 5.4.3. művelet után kapott tömeg az 5.4.2. művelet utáni tömegnél több mint 1 mg-mal kisebb, az 5.4.3. pontban leírtakat megismételjük.
Ha a tömeg növekszik, a számolásnál (6.) a legkisebb értéket vesszük figyelembe.
A véglegesen elfogadott tömeg M2 g lesz.
Az összes szárítási idő általában 6 óránál nem hosszabb.
6. Az eredmény kiszámítása
A minta víztartalmát tömegszázalékban a következő képlettel számítjuk ki:
ahol:
M0 = a bemérőedény és a fedél tömege g-ban az 5.2. művelet után,
M1 = az edény, a fedél és a minta együttes tömege g-ban szárítás előtt (5.3.)
M2 = az edény, a fedél és a minta együttes tömege szárítás után (5.4.3.) vagy (5.4.4.)
7. Ismételhetőség
Párhuzamos vizsgálatok között, ugyanazon mintából, ugyanazon vizsgáló által egy időben vagy közvetlenül egymás után végzett két meghatározás eredménye közötti különbség nem lehet több, mint 0,1 g víz/100 g termék [0,1% (m/m)].
2. módszer: A fehérjetartalom meghatározása
1. Tárgy és alkalmazási terület
Ez a módszer a
– savas kazeinek,
– oltós kazeinek,
– kazeinátok
fehérjetartalmának meghatározására szolgál, kivéve az ammónium-kazeinátot vagy más ammónium- vagy nitrogén vegyületeket tartalmazó kazeinátokat.
2. Fogalommeghatározás
Fehérjetartalom: az ezzel a módszerrel meghatározott nitrogéntartalom 6,38-as faktorral megszorozva, tömegszázalékban kifejezve.
3. A módszer elve
Ismert mennyiségű mintát kálium-szulfát és kénsav elegyével, réz(II)-szulfát katalizátor jelenlétében elroncsolunk, hogy a szerves nitrogént ammóniumsó nitrogénjévé alakítsuk. Az ammóniát desztilláljuk, bórsavoldatban felfogjuk, majd sósav mérőoldattal titráljuk. A nitrogéntartalmat 6,38-as faktorral szorozva fehérje-tartalommá számítjuk át.
4. Vegyszerek
4.1. Kénsav, tömény sűrűsége 1,84 g/ml
4.2. Kálium-szulfát, vízmentes (K2SO4)
4.3. Réz(II)-szulfát-pentahidrát (CuSO4 . 5H2O)
4.4. Szacharóz (C12H22O11)
4.5. Bórsav, 40 g/l oldat
4.6. Nátrium-hidroxid, 30% (m/m) vizes oldat karbonátmentes
4.7. Sósav, 0,1 mol/l
4.8. Indikátorkeverék: 2 g/l, legalább 95% (v/v) etanolban oldott metilvörös oldatot és 1 g/l, legalább 95% (v/v) etanolban oldott metilénkék oldatot azonos térfogatarányban összekeverünk.
5. Eszközök
5.1. Analitikai mérleg, 0,1 mg pontosságú
5.2. Kjeldahl-lombik, 500 ml-es
5.3. Roncsolókészülék, amely a Kjeldahl-lombikot (5.2.) ferde helyzetben tartja olyan melegítőberendezéssel, amellyel elkerülhető, hogy a folyadékfelszín feletti lombikrész felhevüljön.
5.4. Hűtő, egyenes belső csővel
5.5. Biztonsági gömbbel ellátott visszafolyó cső, amely a hűtő alsó részéhez csiszolattal vagy gumicsővel csatlakozik. Amennyiben gumicsatlakozást használunk, az ne legyen túl hosszú.
5.6. Desztilláló feltét, amely a Kjeldahl-lombikhoz (5.2.) és a hűtőhöz (5.4.) puha, jól záró gumiból, vagy más megfelelő anyagból készült dugóval csatlakozik.
5.7. Erlenmeyer-lombik, 500 ml-es
5.8. Mérőhenger, 50 ml-es és 100 ml-es
5.9. Büretta, 50 ml-es, 0,1 ml-es osztással
5.10. Forrást elősegítő anyag
5.10.1. A roncsoláshoz: kis porcelándarabkák vagy üveggyöngyök
5.10.2. A desztillációhoz: forrkő darabkák (csak egyszer használhatók)
6. A vizsgálat menete
6.1. A minta előkészítése
Lásd: I. Minta-előkészítés és általános követelmények 1.2. pont.
6.2. Elővizsgálat az ammónium-nitrogén ellenőrzésére
Ha azt feltételezzük, hogy ammónium-kazeinát vagy más ammónium-vegyület van jelen, hajtsuk végre a következő elővizsgálatot:
1 g mintához egy kis Erlenmeyer-lombikban 10 ml vizet és 100 mg magnézium-oxidot adunk. A lombik belső falára tapadt összes magnézium-oxidot az oldatba beleöblítjük és a lombikot parafadugóval lezárjuk. A dugó és az üvegnyak közé nedves vörös-lakmuszpapír csíkot helyezünk. A lombik tartalmát gondosan összerázzuk, és a lombikot 60–65 °C-os vízfürdőn melegítjük. Ha a lakmuszpapír 15 percen belül kékre színeződik, ammónia van jelen, és a módszer nem alkalmazható (lásd: 1. pont).
6.3. Vakpróba
A minta nitrogéntartalmának meghatározásával egy időben 0,5 g szacharóz jelenlétében vakpróbát is készítünk ugyanazon készülékben, minden reagens ugyanazon mennyiségével és ugyanazzal az eljárással, mint amit a 6.5. pontban leírtunk; ha a vakpróba titrálásra 0,1 mol/l töménységű savból 0,5 ml-nél több fogy, a vegyszereket ellenőrizni kell és a szennyezetteket meg kell tisztítani vagy ki kell cserélni.
6.4. Bemérés
Kjeldahl-lombikba (5.2.) 0,3–0,4 g vizsgálandó mintát (6.1.) 0,1 mg pontossággal bemérünk.
6.5. Meghatározás
6.5.1. A lombikba néhány porcellándarabkát vagy üveggyöngyöt (5.10.1.) és kb. 10 g vízmentes kálium-szulfátot (4.2.) adunk. Ezután hozzáadunk 0,2 g réz(II)-szulfátot (4.3.), a lombik nyakát vízzel utánaöblítjük, végül 20 ml tömény kénsavat (4.1.) és a lombik tartalmát összekeverjük.
A lombikot a roncsolókészüléken (5.3.) lassan melegítjük, amíg a habképződés meg nem szűnik. Lassan erősítjük a hevítést és addig folytatjuk, amíg az oldat már egyetlen fekete részecskét sem tartalmaz és halvány zöldeskék színű nem lesz. A lombikot melegítés közben időnként óvatosan összerázzuk.
A forralást úgy kell szabályozni, hogy a gőzök a lombik nyakának közepén kondenzálódjanak. A forralást a helyi túlmelegedést elkerülve 90 pecig folytatjuk. Végül a lombikot tartalmával együtt szobahőmérsékletre hűtjük. Azután óvatosan hozzáadunk 200 ml vizet és néhány darab forrkövet (5.10.2.), összekeverjük és újra lehűtjük.
6.5.2. Az Erlenmeyer-lombikba (5.7.) 50 ml bórsavoldatot (4.5.) és 4 csepp indikátort (4.8.) adunk és tartalmát összekeverjük. A lombikot a hűtő (5.4.) alá csatlakoztatjuk úgy, hogy a lefolyócső vége (5.5.) a bórsavoldatba merüljön. Mérőhengerrel (5.8.) 80 ml nátrium-hidroxid oldatot (4.6.) töltünk a Kjeldahl-lombikba. Ezalatt a lombikot megdöntve tartjuk úgy, hogy a nátrium-hidroxid oldat a lombik belső falán végigfolyva alsó réteget képezzen. Ezután a Kjeldahl-lombikot azonnal a feltét (5.6.) segítségével a hűtőhöz csatlakoztatjuk. A Kjeldahl-lombikot óvatosan megforgatjuk, hogy tartalma összekeveredjen. A felhabzás elkerülésére lassan melegítjük.
A desztillációt 150 ml desztillátum összegyűléséig – kb. 30 percen keresztül – folytatjuk. A desztillátum hőmérséklete 25 °C alatt legyen. Kb. 2 perccel a desztilláció befejezése előtt az Erlenmeyer-lombikot úgy süllyesztjük, hogy a cső vége már ne érjen bele a savoldatba, a cső végét kevés vízzel leöblítjük. A melegítést befejezzük, eltávolítjuk a csövet úgy, hogy mind a belső, mind a külső falát kevés vízzel leöblítjük és az öblítővizet az Erlenmeyer-lombikban felfogjuk.
6.5.3. Az Erlenmeyer-lombikban felfogott desztillátumot 0,1 mol/l töménységű sósavoldattal (4.7.) megtitráljuk.
7. Az eredmények kiszámítása
A minta fehérjetartalmát tömegszázalékban kifejezve a következő képlettel határozzuk meg:
|
(V1–V2) ⋅ T ⋅ 14 ⋅ 100 ⋅ 6,38 |
= |
8,932 ⋅ (V1–V2) ⋅ T |
|
m ⋅ 1000 |
m |
ahol:
V1 = a meghatározáshoz (6.5.) fogyott sósav mérőoldat (4.7.) térfogata ml-ben
V2 = a vakpróbára (6.3.) fogyott sósav mérőoldat (4.7.) térfogata ml-ben
T = a sósav mérőoldat (4.7.) faktora
m = a bemért minta tömege g-ban
A fehérjetartalmat 0,1% (m/m) pontossággal kell megadni.
8. Ismételhetőség
Párhuzamos, azaz ugyanazon vizsgáló által ugyanazon mintából azonos időben, vagy közvetlenül egymás után végzett vizsgálatok között a megengedett eltérés legfeljebb 0,5 g fehérje/100 g termék [0,5% (m/m)].
3. módszer: A titrálható savasság meghatározása
1. Tárgy és alkalmazási terület
A módszer a savkazein titrálható savasságának meghatározására szolgál.
2. Fogalommeghatározás
Titrálható savasság: 0,1 mol/l töménységű nátrium-hidroxid mérőoldat ml-ben mért térfogata, amely a termék 1 g-ja vizes extraktumának közömbösítéséhez szükséges.
3. A módszer elve
A minta 60 °C-on kivont vizes extraktumát szűrjük. A szűrletet nátrium-hidroxid mérőoldattal fenolftalein indikátor jelenlétében megtitráljuk.
4. Vegyszerek
Az eljáráshoz vagy a reagensekhez szükséges vizet előzetesen 10 percen át forralva szén-dioxid-mentesíteni kell.
4.1. Nátrium-hidroxid mérőoldat, 0,1 mol/l
4.2. Fenolftalein indikátor 10 g/l, etanolos oldat 95% (v/v), közömbösített
5. Eszközök
5.1. Analitikai mérleg, 0,1 mg pontosságú
5.2. Erlenmeyer-lombik, 500 ml-es, becsiszolt dugóval
5.3. Pipetta, 100 ml-es
5.4. Pipetta, 0,5 ml indikátoroldat (4.2.) adagolására
5.5. Erlenmeyer-lombik, 250 ml-es
5.6. Mérőhenger, 250 ml-es
5.7. Büretta, 0,1 ml osztással
5.8. Vízfürdő, hőmérséklete 60±2 °C-ra szabályozható
5.9. Megfelelő szűrő
6. A vizsgálat menete
6.1. A vizsgálandó minta előkészítése
(Lásd: az I. Minta-előkészítés és általános követelmények 1.2. pont)
6.2. Bemérés
Kb. 10 g mintát (6.1.) 10 mg pontossággal lemérünk és Erlenmeyer-lombikba (5.2.) tesszük.
6.3. Meghatározás
250 ml-es mérőhengerrel (5.6.) 200 ml frissen kiforralt és lehűtött, majd 60 °C-ra melegített vizet Erlenmeyer-lombikba mérünk. A lombikot lezárjuk, rázással összekeverjük és 60 °C-ra beállított vízfürdőben (5.8.) 30 percen keresztül melegítjük. A lombikot 10 perces időközönként összerázzuk. Szűrjük és a szűrletet 20 °C-ra lehűtjük. A szűrlet tiszta legyen. A lehűtött szűrlet 100 ml-ét pipettával (5.3.) Erlenmeyer-lombikba (5.5.) mérjük. Pipettával (5.4.) hozzáadunk 0,5 ml fenolftalelein indikátor-oldatot (4.2.). Addig titráljuk a nátrium-hidroxid mérőoldattal (4.1.), amíg halvány rózsaszínű nem lesz, és a szín legalább 30 másodpercig meg is marad. A fogyott mennyiséget 0,01 ml pontossággal leolvassuk és feljegyezzük.
7. Az eredmény kiszámítása
A kazein titrálható savasságát a következő képlettel számítjuk ki:
20 ⋅ V ⋅ T
m
ahol:
V = a fogyott normál nátrium-hidroxid oldat (4.1.) térfogata, ml-ben,
T = a nátrium-hidroxid oldat faktora (4.1.), mol/l,
m = a bemért minta tömege, g-ban.
A titrálható savasságot két tizedes pontossággal fejezzük ki.
8. Ismételhetőség
A párhuzamos, azaz ugyanazon vizsgáló által ugyanazon mintából, azonos időben, vagy közvetlenül egymás után végzett vizsgálatok között a megengedett eltérés legfeljebb 0,02 ml 0,1 mol/l nátrium-hidroxid mérőoldat/1 g termék.
4. módszer: A hamutartalom meghatározás (P2O5-ot beleértve)
1. Tárgy és alkalmazási terület
A módszer a savas kazein hamutartalmának (P2O5-ot beleértve) meghatározására szolgál.
2. Fogalommeghatározás
Hamutartalom (P2O5-ot beleértve): a következőkben leírt módszerrel meghatározott hamutartalom.
3. A módszer elve
A vizsgálandó mintát 825±25 °C-on az összes szerves eredetű foszfort megkötő magnézium-acetát jelenlétében elhamvasztjuk. A hamutartalmat a maradék és a magnézium-acetátból származó hamu mennyiség különbsége adja.
4. Vegyszerek
4.1. Magnézium-acetát-tetrahidrát oldat, 120 g/l
A magnézium-acetát-tetrahidrát (Mg(CH3CO2)2 . 4H2O) 120 g-ját vízben oldjuk, és 1 literre feltöltjük.
5. Eszközök
5.1. Analitikai mérleg, 0,1 mg pontosságú
5.2. Pipetta, 5 ml-es
5.3. Izzítótégely, kvarcból vagy platinából, átmérője kb. 70 mm, magassága 25–50 mm
5.4. Szárítószekrény, 102±1 °C-ra beállítható
5.5. Izzítókemence, 825±25 °C-ra beállítható
5.6. Vízfürdő, forrásban tartható
5.7. Exszikkátor, nedvességre érzékeny indikátort tartalmazó frissen aktívált szilikagéllel vagy azonos hatékonyságú szárítóközeggel.
6. A vizsgálat menete
6.1. A vizsgálandó minta előkészítése
Lásd: I. Minta-előkészítés és általános követelmények 1.2. pont.
6.2. Az izzítótégely előkészítése
Két izzítótégelyt a 825±25 °C-ra beállított izzítókemencében (5.5.) 30 percig hevítünk. Azután az izzítótégelyeket exszikkátorban (5.7.) a mérlegszoba hőmérsékletére hűtjük és 0,1 mg pontossággal mérjük.
6.3. Bemérés
A fenti módon előkészített izzítótégelybe (A) kb. 3 g vizsgálandó mintát (6.1.) 0,1 mg pontossággal bemérünk.
6.4. Meghatározás
Az izzítótégelybe (A) pipettával (5.2.) pontosan 5 ml magnézium-acetát oldatot (4.1.) adunk úgy, hogy az egész minta átnedvesedjen, ezután 20 percig állni hagyjuk.
Egy másik előkészített izzítótégelybe (B) pipettával (5.2.) pontosan 5 ml magnézium-acetát oldatot (4.1.) adunk. Mindkét izzítótégely (A és B) tartalmát forrásban lévő vízfürdőn (5.6.) szárazra pároljuk.
Mindkét izzítótégelyt 30 percre a 102±1 °C-ra beálllított szárítószekrénybe (5.4.) helyezzük. Az A jelű izzítótégely tartalmát kis lánggal melegített forró lapon vagy infravörös lámpa alatt addig hevítjük, amíg a vizsgálandó anyagmennyiség teljesen el nem szenesedik. Ügyeljünk arra, hogy a tartalma lángra ne lobbanjon.
Mindkét tégelyt (A és B) 825±25 °C-ra beállított izzítókemencébe (5.5.) tesszük és legalább 1 óra hosszat a kemencében hagyjuk, míg az A tégelyben elszenesedett részeket már nem találunk. Mindkét izzítótégelyt exszikkátorban (5.7.) a mérlegszoba hőmérsékletére hűtjük, majd 0,1 mg pontossággal mérjük.
Az elektromos izzítókemencében (5.5.) történő hevítést a lehűtést és a mérést mindaddig ismételjük, amíg a tömeg 1 mg pontossággal állandó marad, vagy növekedni nem kezd. A legkisebb tömeget jegyezzük fel.
7. Az eredmény kiszámítása
A hamutartalmat (P2O5-ot beleértve) tömegszázalékban kifejezve a következő képlettel számítjuk:
|
(m1 – m2) – (m3 – m4) |
⋅ 100 |
|
m0 |
ahol:
m0 = a bemért minta tömege g-ban,
m1 = az A izzítótégely és a maradék tömege g-ban,
m2 = az előkészített A tégely tömege g-ban,
m3 = a B izzítótégely és a maradék tömege g-ban,
m4 = az előkészített B izzítótégely tömege g-ban.
Az eredményeket 0,01% (m/m) pontossággal adjuk meg.
8. Ismételhetőség
Ugyanabból a mintából, azonos vizsgáló által, azonos körülmények között egyidőben vagy közvetlenül egymás után végzett két meghatározás eredménye között a különbség legfeljebb 0,1 g/100 g termék [0,1% (m/m)] lehet.
5. módszer: A hamutartalom meghatározása (P2O5-ot is beleértve)
1. Tárgy és alkalmazási terület
Ez a módszer oltós kazeinben lévő hamutartalom meghatározására szolgál.
2. Fogalommeghatározás
A hamutartalom (P2O5-ot beleértve): ezzel a módszerrel meghatározott hamutartalom.
3. A módszer elve
A minta ismert mennyiségét 825±25 °C-on tömegállandóságig hamvasztjuk. A maradékot mérjük és az eredményt a minta tömegszázalékában adjuk meg.
4. Eszközök
4.1. Analitikai mérleg, 0,1 mg pontosságú
4.2. Izzítótégely kvarcból vagy platinából, átmérője kb. 70 mm, magassága 25–50 mm
4.3. Elektromos izzítókemence 825±25 °C-ra szabályozható
4.4. Exszikkátor, nedvességre érzékeny indikátort tartalmazó frissen aktivált szilikagéllel, vagy azonos hatékonyságú szárítóközeggel
5. A vizsgálat menete
5.1. A vizsgálandó minta előkészítése
Lásd: I. Minta-előkészítés és általános körülmények (1.2. pont).
5.2. Az izzítótégely előkészítése
Az izzítótégelyt a 825±25 °C-ra beállított elektromos izzítókemencében (4.3.) 30 percen keresztül izzítjuk. Ezután exszikkátorban (4.4.) a mérlegszoba hőmérsékletére hűtjük és 0,1 mg pontossággal mérjük.
5.3. Bemérés
A fenti módon előkészített izzítótégelybe a vizsgálandó mintából (5.1.) közvetlenül vagy visszaméréssel kb. 3 g-ot 0,1 mg pontossággal bemérünk.
5.4. Meghatározás
Az izzítótégelyt tartalmával együtt kis lángon vagy infravörös lámpával a mintamennyiség teljes elszenesedéséig hevítjük. Ügyeljünk arra, hogy a tartalom lángra ne lobbanjon.
Ezután az izzítótégelyt 825±25 °C hőmérsékletre beállított elektromos izzítókemencébe (4.3.) tesszük és legalább 1 óra hosszat, az izzítótégelyben lévő szénrészecskék teljes eltűnéséig hevítjük.
Ezután az izzítótégelyt exszikkátorban (4.4.) a mérleg-szoba hőmérsékletére hűtjük és 0,1 mg pontossággal mérjük. Az izzítást, a lehűtést és a mérést addig ismételjük, amíg a tömeg 1 mg pontossággal állandó marad, vagy növekedni nem kezd. A legkisebb tömeget jegyezzük fel.
6. Az eredmény kiszámítása
A minta hamutartalmát tömegszázalékban a következő képlettel számítjuk ki:
ahol:
m0 = a bemért minta tömege mg-ban,
m1 = az izzítótégely és a maradék tömege mg-ban,
m2 = az izzítótégely tömege mg-ban.
Az eredményeket 0,01% (m/m) pontossággal adjuk meg.
7. Ismételhetőség
Ugyanabból a mintából, azonos vizsgáló által, azonos körülmények között egyidőben vagy közvetlenül egymás után végzett két meghatározás eredménye között a a különbség legfeljebb 0,15 g/100 g termék [0,15% (m/m)] lehet.
6. módszer: A pH-érték meghatározása
1. Tárgy és alkalmazási terület
Ez a módszer a kazeinát pH-értékének meghatározására szolgál.
2. Fogalommeghatározás
A kazeinát pH-értéke: a kazeinát vizes oldatának 20 °C-on ezzel a módszerrel meghatározott pH-értéke.
3. A módszer elve
A kazeinát vizes oldata pH-értékének elektrometriás meghatározása pH-mérővel.
4. Vegyszerek
A vegyszerek elkészítéséhez vagy a vizsgálathoz (6.) felhasznált vizet közvetlenül a felhasználás előtt desztillálni és a széndioxid-abszorpció ellen védeni kell.
4.1. Puffer-oldat a pH-mérő (5.2.) kalibrálásához
Két standard puffer-oldatot használunk, amelyeknek pH-értéke 20 °C-on két tizedesjegy pontossággal ismert. Az egyik pH-értéke kisebb, a másiké nagyobb legyen, mint a vizsgálandó mintáé. Például 4 körüli pH-jú ftalát-puffer-oldatot és 9 körüli pH-jú bórax-puffer-oldatot használhatunk.
5. Eszközök
5.1. Mérleg, leolvasási pontossága 0,1 mg
5.2. pH-mérő, leolvasási pontossága 0,05 pH egység, megfelelően hitelesített üveg-, kalomel vagy más vonatkoztatási elektróddal
5.3. Hőmérő, leolvasási pontossága 0,5 °C
5.4. Erlenmeyer-lombik, 100 ml-es, becsiszolt üvegdugóval
5.5. Főzőpohár, 50 ml-es
5.6. Keverő
5.7. Főzőpohár a keveréshez (5.6.), térfogata legalább 250 ml
6. A vizsgálat menete
6.1. A vizsgálandó minta előkészítése
Lásd: I. Minta-előkészítés és általános követelmények (1.2. pont).
6.2. Meghatározás
6.2.1. A mérőműszer hitelesítése
A puffer-oldatok (4.1.) hőmérsékletét 20 °C-ra állítjuk be és a pH-mérőt a használati utasításnak megfelelően hitelesítjük.
Megjegyzések:
1. A hitelesítést addig kell elvégezni, amíg a vizsgálandó oldatot 20 percig állni hagyjuk (6.2.2.).
2. Ha mintasorozatot vizsgálunk, a pH-mérő egy vagy több standard oldattal végzett hitelesítését legalább 30 percenként meg kell ismételni.
6.2.2. A vizsgálandó oldat előkészítése
A főzőpohárba (5.7.) 95 ml vizet engedünk, hozzáadunk 5 g vizsgálandó mintát (6.1.) és a keverővel (5.6.) 30 másodpercig keverjük. 20 percig 20 °C-on állni hagyjuk; közben a főzőpoharat óraüveggel lefedjük.
6.2.3. A pH-érték meghatározása
Az oldatból körülbelül 20 ml-t a főzőpohárba (5.5.) viszünk és a pH-mérővel (5.2.) az oldat pH-értékét azonnal mérjük. A pH-mérő üvegelektródját előzőleg vízzel gondosan öblítsük le. A kazeinát vizes oldatának pH-értékét a pH-mérő skáláján legalább 2 tizedes pontossággal leolvassuk és feljegyezzük.
7. Ismételhetőség
Ugyanabból a mintából, azonos vizsgáló által, azonos körülmények között egyidőben vagy közvetlenül egymás után végzett két meghatározás eredménye között a különbség legfeljebb 0,05 pH-érték lehet.
MAGYAR ÉLELMISZERKÖNYV
(Codex Alimentarius Hungaricus)
3–1–86/424 számú előírás
(2. kiadás – 2006.)
Az étkezési kazeinek és kazeinátok mintavételi módszerei
Az étkezési kazeinek és kazeinátok mintavételi módszereit és az ehhez szükséges eszközöket az előírás 1. számú melléklete tartalmazza. Ez az előírás 2007. július 1-jén lép hatályba, ezzel egyidejűleg a Magyar Élelmiszerkönyvnek az étkezési kazeinek és kazeinátok mintavételi módszereiről szóló 3–1–86/424 számú előírása 1995-ben jóváhagyott 1. kiadása hatályát veszti.
Ez az előírás az étkezési kazeinek és kazeinátok vegyi elemzéséhez szükséges közösségi mintavételi módszerek megállapításáról szóló, 1986. július 15-i 86/424/EGK bizottsági irányelvnek való megfelelést szolgálja.
1. számú melléklet a 3–1–86/424 számú előíráshoz
Egyes emberi fogyasztásra szánt étkezési kazeinek és kazeinátok mintavételi módszerei
I. Általános előírások
1. Adminisztratív előírások
1.1. A mintavevőre vonatkozó előírás
A mintavételt az érvényes előírásoknak megfelelően képzett és meghatalmazott személy végezze.
1.2. A minták lezárása és jelölése
Minden hivatalos mintát a vétel helyszínén le kell zárni, le kell pecsételni és az azonosításhoz szükséges adatokat fel kell tüntetni rajta.
1.3. Párhuzamos minták
A vizsgálatokhoz legalább két azonos, reprezentatív mintát kell egyidejűleg venni. A mintákat a mintavétel után a lehető leghamarabb el kell küldeni a laboratóriumba.
Az egyik minta ellenmintaként szolgál.
1.4. Mintavételi jegyzőkönyv
A mintához mintavételi jegyzőkönyvet kell mellékelni. A jegyzőkönyvet a mintavétellel megbízott és tanúként jelen lévő személyek írják alá. A jegyzőkönyvnek a következőket kell tartalmaznia:
a) a mintavétel helyét, dátumát, időpontját,
b) a mintavevő személy és a tanú nevét,
c) a mintavétel pontos leírását,
d) a szállítmányt alkotó tételek fajtáját, számát, kódját,
e) annak a tételnek az azonosítási számát, amelyből a mintát vették,
f) a tételből vett minták számát, utalva arra a tételre, amelyből a minták származnak,
g) a helyet, ahová a mintát küldeni kell,
h) a gyártó vagy a csomagolásért felelős személy nevét és címét.
2. Mintavevő eszközök
Minden eszköz legyen alkalmas a mintavételre, és ne okozzon a mintában semmilyen, a vizsgálat eredményét befolyásoló változást. Rozsdamentes acélból készült eszközök használata ajánlott.
Minden felület sima, karcmentes, minden sarok legömbölyített legyen. A mintavevő eszközök feleljenek meg az egyes vizsgálandó termékekre érvényes követelményeknek.
3. Mintatároló edények
A mintatároló edények és fedelük olyan anyagból, olyan kialakítással készüljenek, hogy védjék a mintát minden olyan lehetséges változástól, ami a vizsgálatok eredményét befolyásolhatja. Az alkalmas anyagok közé tartozik az üveg, egyes fémek és egyes műanyagok. Az edények átlátszatlansága előnyös. Ha fényáteresztő edényeket használunk, mintával megtöltve sötét helyen tároljuk. Az edények és a fedők tiszták és szárazak legyenek.
Felhasználhatók eldobható műanyag edények, alumíniumfóliával rétegezett műanyagból készült edények és megfelelő zárással ellátott alkalmas műanyag zacskók. A műanyag zacskókon kívül a többi edény légmentesen zárható legyen, vagy alkalmas dugóval, vagy, szükség esetén, nedvességet szigetelő, nem oldódó, nem abszorbeáló és zsírnak ellenálló műanyag bevonattal ellátott menetes fém/műanyag kupakkal azért, hogy a minta szagának, ízének, tulajdonságainak vagy összetételének bármilyen változását elkerüljük. Ha dugót használunk, az nem abszorbeáló, szagmentes anyagból készüljön.
4. A mintavétel technikája
A mintatároló edényt a mintavétel után azonnal zárjuk le.
5. A minták tárolása
A tárolási hőmérséklet a különböző kazein- és kazeinát-minták elszállítását megelőzően ne haladja meg a 25 °C-ot.
6. A minták szállítása
A mintákat a mintavétel után a lehető leggyorsabban (lehetőség szerint 24 órán belül) el kell juttatni a vizsgáló laboratóriumba. A szállítás alatt gondoskodni kell arról, hogy a légnemű szennyeződések, a közvetlen napfény és a 25 °C feletti hőmérséklet okozta károsodásokat elkerüljük.
II. Étkezési kazeinek és kazeinátok mintavételi módszerei
1. Tárgy és alkalmazási terület
Ezek a módszerek
– étkezési savas kazeinek,
– étkezési oltós kazeinek,
– étkezési kazeinátok
mintavételére szolgálnak.
2. Eszközök
Lásd: Általános előírások, 2. pont.
2.1. Szúró mintavevők, amelyek elegendő hosszúak ahhoz, hogy a terméket tartalmazó tartály aljáig leérjenek. Ezek a szúró mintavevők ezen előírás III. részében találhatók.
2.2. Kanál vagy széles végű spatula
2.3. Mintatároló edények
Lásd: Általános előírások, 3. pont.
3. Mintavételi eljárás
3.1. Általános előírások
Mindent el kell követni, hogy a vizsgálandó minta vétele előtt és alatt az edény és a minta által felvett légnedvesség a lehető legkisebb legyen. Az edényt a mintavétel után újból gondosan zárjuk le.
3.2. Mintavétel
3.2.1. Mintavétel kémiai vizsgálathoz
A kivett minta mennyisége legalább 200 g legyen. A tiszta és száraz szúró mintavevőt a terméken keresztülvezetjük, szükség esetén a tartályt megdöntjük vagy oldalára fektetjük. A szúró mintavevő nyílását lefelé irányítjuk, a bevezetés egyenletes legyen. Ha a szúró mintavevő a tartály alját elérte, 180°-kal elfordítjuk, kihúzzuk és tartalmát a mintatároló edénybe töltjük. A 200 g-os mennyiséghez egy vagy több mintavétel szükséges. Mihelyt összegyűlt az elegendő minta mennyiség, a mintatároló edényt azonnal lezárjuk.
3.2.2. Mintavétel fogyasztói csomagokból
A mintavételhez sértetlen, bontatlan fogyasztói csomagot használunk. Ugyanazon árutételből vagy azonos számmal jelzett csomagokból egy vagy több csomagot veszünk ki ahhoz, hogy legalább 200 g-os mintát nyerjünk.
Ha az instant tulajdonságok meghatározása szükséges, mindig ezt a mintavételi eljárást alkalmazzuk.
3.2.3. A minta megőrzése, tárolása és szállítása
Lásd: Általános előírások, 5. és 6. pont.
III. Szúró mintavevők étkezési kazeinek és kazeinátok mintavételéhez
1. Szúró mintavevő fajták
A típus: hosszú
B típus: rövid
(Lásd az ábrát)
2. A mintavevő eszközök leírása
A hengeres test és a fogantyú polírozott fémből, lehetőleg rozsdamentes acélból készüljön. A hosszú szúró mintavevő markolata rozsdamentes acélból készüljön. A rövid szúró mintavevő markolata levehető, fából vagy műanyagból készült, a tulajdonképpeni szúró mintavevőhöz bajonettzárral csatlakozik.
3. A mintavevő eszköz megmunkálása
3.1. Az eszköz alakja, anyaga és megmunkálása olyan legyen, hogy az könnyen tisztítható és szükség esetén sterilezhető legyen.
3.2. Az A típusú szúró mintavevő hengeres testének széle elegendően éles legyen ahhoz, hogy kaparóként szolgáljon.
3.3. A szúró mintavevő hegye elég éles legyen, hogy a mintavételt megkönnyítse.
4. Fő méretek
A szúró mintavevők feleljenek meg a következő táblázatban feltüntetett méreteknek, 10% tűréssel:
|
Méretek milliméterben |
|
A típus
hosszú |
B típus
rövid |
A szúró mintavevő hossza |
800 |
400 |
A fémlap vastagsága |
1–2 |
1–2 |
A szúró mintavevő belső átmérője a csúcsnál |
18 |
32 |
A szúró mintavevő belső átmérője a nyél alatt |
22 |
28 |
Résszélesség a csúcsnál |
4 |
20 |
Résszélesség a nyél alatt |
14 |
14 |
5. Használati utasítás a szúró mintavevőkhöz
5.1. Többé-kevésbé könnyen folyó porokba a szúró mintavevők függőlegesen vezethetők be. Az A típusú szúró mintavevő forgatással teljesen megtölthető és függőlegesen visszahúzható. A B típusú szúró mintavevő a bevezetés alatt már teljesen megtelik, visszahúzáskor azonban megfelelő helyzetben kell tartani, hogy a veszteséget az alsó végén elkerüljük.
5.2. Többé-kevésbé szabadon folyó poroknál a tartályt megdöntjük és a szúró mintavevőt majdnem vízszintesen, réssel lefelé vezetjük be, majd réssel felfelé húzzuk ki.
Ábra
Szúró mintavevő kazeinek és kazeinátok mintavételére
(méretek mm-ben)
MAGYAR ÉLELMISZERKÖNYV
(Codex Alimentarius Hungaricus)
3–1–87/524 számú előírás
(2. kiadás – 2006.)
Sűrített tej és tejporfélék mintavételi módszerei
Az étkezési sűrített tej és a tejpor vizsgálatához szükséges mintavételt a mellékletben leírtak szerint kell elvégezni.
Ez az előírás 2007. július 1-jén lép hatályba, ezzel egyidejűleg a Magyar Élelmiszerkönyvnek a sűrített tej és tejporfélék mintavételi módszereiről szóló 3–1–87/524 számú előírása 1995-ben jóváhagyott 1. kiadása hatályát veszti.
Ez az előírás a tartós tejtermékek vizsgálatára szolgáló vegyi elemzéshez szükséges közösségi mintavételi módszerek megállapításáról szóló, 1987. október 6-i 87/524/EGK bizottsági irányelvnek való megfelelést szolgálja.
Melléklet a 3–1–87/524 számú előíráshoz
I. Általános előírások
1. Adminisztratív előírások
1.1. A mintavevőre vonatkozó előírás
A mintavételt az érvényes előírásoknak megfelelően képesített és felhatalmazott személy végezze.
1.2. A minták lezárása és jelölése
Minden hivatalos mintát a vétel helyszínén le kell pecsételni és az előírások szerint megjelölni.
1.3. Párhuzamos minták
A vizsgálatokhoz legalább két azonos mintát kell egyidejűleg venni. Az egyik minta ellenmintaként szolgál.
A mintákat a mintavétel után a lehető leghamarabb el kell küldeni a laboratóriumba.
1.4. Jegyzőkönyv
A mintához mintavételi jegyzőkönyvet kell mellékelni.
A jegyzőkönyvet a mintavétellel megbízott és tanúként jelen lévő személyek írják alá. A jegyzőkönyvnek a következőket kell tartalmaznia:
a) a mintavétel helye, dátuma, időpontja,
b) a mintavevő személy és a tanú neve,
c) a mintavétel pontos leírása,
d) a szállítmányt alkotó tételek fajtája, száma, kódja,
e) annak a tételnek az azonosítási száma, amiből a mintát vették,
f) a tételből vett minták száma, utalva arra a tételre, amiből a minták származnak,
g) a hely, ahová a mintát küldeni kell,
h) a gyártó vagy a csomagolásért felelős személy neve és címe.
2. Mintavevő eszközök
Minden eszköz legyen alkalmas a mintavételre, és ne okozzon a mintában semmilyen, a vizsgálat eredményét befolyásoló változást. Rozsdamentes acélból készült eszközök használata ajánlott, melyek felülete sima, karcmentes, minden sarok legömbölyített legyen.
A mintavevő eszközök feleljenek meg az egyes vizsgálandó termékekre érvényes követelményeknek.
3. Mintatároló edények
A mintatároló edényeket és fedelüket olyan anyagból, olyan kialakítással kell készíteni, hogy védjék a mintát minden olyan lehetséges változástól, ami a vizsgálatok eredményét befolyásolhatja. Az alkalmas anyagok közé tartozik az üveg, egyes fémek és egyes műanyagok. Az edények átlátszatlansága előnyös. Ha fényáteresztő edényeket használunk, a mintával megtöltve sötét helyen tároljuk azokat.
Az edények és a fedők tiszták és szárazak legyenek. Az edények alakja és térfogata feleljen meg a vizsgálandó termékre érvényes követelményeknek.
Felhasználhatók eldobható műanyag edények, alumíniumfóliával rétegezett műanyagból készült edények és megfelelő zárással ellátott alkalmas műanyag zacskók. A műanyag zacskókon kívül a többi edény légmentesen zárható legyen vagy erre alkalmas dugóval, vagy szükség esetén nedvességet szigetelő, nem oldódó, nem abszorbeáló, zsírnak ellenálló műanyag bevonattal ellátott, menetes fém/műanyag kupakkal, azért hogy a minta szagának, ízének, tulajdonságainak vagy összetételének bármilyen változását elkerüljük.
Ha dugót használunk, az nem abszorbeáló, szagmenetes anyagból készüljön.
4. A mintavétel technikája
A mintatároló edényt a mintavétel után azonnal zárjuk le.
5. A minták tárolása
A tárolási hőmérséklet a minták elszállítását megelőzően ne haladja meg a 25 °C-ot.
6. A minták szállítása
A mintákat a mintavétel után a lehető leggyorsabban (lehetőség szerint 24 órán belül) el kell juttatni a vizsgáló laboratóriumba. A szállítás alatt gondoskodni kell arról, hogy a légnemű szennyeződések, a közvetlen napfény és a 25 °C feletti hőmérséklet okozta károsodásokat elkerüljük.
II. 1. módszer: Mintavétel sűrített tejből
1. Tárgy és alkalmazási terület
A módszert a következő tejfélék kémiai vizsgálatához szükséges minta vételére alkalmazzuk:
– cukrozatlan sűrített tej,
– cukrozatlan sűrített soványtej,
– cukrozatlan sűrített félzsíros tej,
– cukrozatlan sűrített zsíros tej,
– cukrozott sűrített tej,
– cukrozott sűrített soványtej,
– cukrozott sűrített félzsíros tej.
2. Eszközök
2.1. Általános megjegyzések
Lásd: Általános előírások, 2. pont.
2.2. Perforált tárcsás és egyéb keverők
A keverők a nagy mennyiségű folyadékok átkeveréséhez, örvénylő mozgás keltéséhez elegendő nagy felületűek legyenek, azonban ne segítsék elő avas íz és szag kifejlődését.
A tartályok különféle formája és nagysága miatt nem létezik minden célra megfelelő keverő. A keverőt úgy kell kialakítani, hogy a tartály belső felületét a keverés során ne karcolja fel. Az eszközöket az Általános előírások 2. pontja írja le. Vödörben, kannában lévő folyadékok keverésére alkalmas keverő (lásd: 1. ábra) tájékoztató méretei: 150 mm átmérőjű tárcsa, 100 mm átmérőjű kör mentén elhelyezkedő hat, egyenként 12,5 mm átmérőjű lyukkal. A középpontban olyan nem-fém rúd csatlakozik, amelynek a másik végén hurokszerű fogantyú van. A rúd hossza fogantyúval együtt kb. 1 méter.
Kis tartályokhoz alkalmas keverő közelítő méretei (lásd: 2. ábra): legalább 2 m hosszú rúdhoz 300 mm átmérőjű tárcsa csatlakozik; a tárcsán 230 mm átmérőjű kör mentén 12, egyenként 30 mm átmérőjű lyuk található. Nagy tartályok mechanikus keveréséhez tiszta sűrített levegő ajánlott. A lehető legkisebb légnyomást és a legkisebb levegőmennyiséget alkalmazzuk az avas íz és szag kifejlődésének elkerülésére.
Megjegyzés: A tiszta sűrített levegőn az összes szennyeződéstől (beleértve az olajat, vizet és port) megtisztított levegőt értjük.
2.3. Keverő
A keverőnek széles lapátja legyen és érjen le az edény aljáig. A keverő egyik oldala pontosan illeszkedjék a tartály alakjához (lásd: 3. ábra).
2.4. Merítőkanál
Mintavételre alkalmas méretű és formájú merítőkanalat mutat be a 4. ábra. A merítőkanálnak legalább 150 mm hosszú merev nyele legyen, térfogata pedig legalább 50 ml. Előnyös, ha a nyél nincs meghajlítva. A kanalak újabb formája lehetővé teszi egymásba illesztett tárolásukat.
Alkalmazható még az előbbivel azonos térfogatú, jellel öt azonos térfogatszakaszra osztott, hengeres merítőkanál is, amely több edényben érkező áru mintavételét könnyíti meg.
2.5. Keverő rúd
Hengeres, kb. 1 m hosszú, 35 mm átmérőjű.
2.6. Edény
Részminták vételéhez, 5 l térfogatú, szélesszájú.
2.7. Kanál vagy spatula
A spatula lapát része széles legyen.
2.8. Mintatároló edény
Lásd: Általános előírások, 3. pont.
3. Eljárás
3.1. Mintavétel cukrozatlan sűrített tejből.
Legalább 200 g mintát kell venni.
3.1.1. Az edény tartalmát egy keverőrúd le-fel mozgatásával, keverővel való keveréssel, mechanikus mozgatással egyik edényből a másikba való áttöltéssel vagy tiszta sűrített levegővel (lásd: 2.2. pont) alaposan átkeverjük a kielégítően homogén állapot elérése céljából. A mintát közvetlenül a keverés után merítőkanállal vesszük ki. Ha a megfelelő homogén állapot nehezen érhető el, a mintát az edény különböző részeiből kell kivenni. Az összes mintamennyiség legalább 200 g legyen.
Ha a minta részminták elegye, azt a minta címkéjén vagy a kísérő jegyzőkönyvben fel kell tüntetni.
3.1.2. Mintavétel fogyasztói csomagolásból
Sértetlen, bontatlan csomagot lehetőleg azonos gyártásból származó vagy azonosan megjelölt csomagokat kell kivenni. A minta összes mennyisége legalább 200 g legyen.
3.2. Mintavétel cukrozott sűrített tejből
3.2.1. Általános előírások
Nagy tartályokban lévő cukrozott sűrített tejből a mintavétel nagy nehézséggel járhat, különösen akkor, ha a termék inhomogén és nagy viszkozitású. A mintavételnél gondot okozhatnak az egész termékben megjelenő vagy a falakon tapadó, csomósodást okozó szacharóz vagy laktóz kristályok, sókiválások. Ezek a körülmények felismerhetők, ha mintavevőt vezetünk be a tartályba, és azzal a tartály tartalmát megmozgatjuk.
Ha a cukorkristályok mérete 6 mm-nél kisebb, a mintavétel nem okoz gondot. Ha a termék nem homogén, azt a mintán és a csatolt jegyzőkönyvben fel kell tüntetni. Mivel a cukrozott sűrített tejet gyakran a külső hőmérséklettel azonos hőmérsékleten tárolják, a reprezentatív minta kivételéhez tanácsos a termék hőmérsékletét legalább 20 °C-ra emelni.
3.2.2. Eljárás
Legalább 200 g-os mintát kell venni.
– Nyitható tartályok.
A tartály egyik fedőlapját kinyitás előtt kívülről alaposan megtisztítjuk és megszárítjuk annak elkerülésére, hogy a nyitás során idegen anyagok jussanak a tartályba. Tartalmát keverő (lásd: 3. ábra) segítségével jól átkeverjük. A keverő élével a fal belső oldalára és az aljára tapadó terméket lekaparjuk. A ferdén tartott keverő forgó és függőleges mozgatásával a tartályt jól átkeverjük, ügyelve arra, hogy levegőt ne juttassunk bele. A keverőt kivesszük, a rátapadt sűrített tejet spatulával vagy kanállal az 5 literes edénybe (2.6.) kaparjuk. A keverést és a keverő kiemelését addig ismételjük, míg 2-3 l össze nem gyűlik, majd a homogénné kevert sűrített tejből legalább 200 g-ot kiveszünk.
– Zárt tartályok (dobok), peremes nyílással a fedőlapon vagy a paláston.
A 3.2.1. pontban leírt okokból a mintavétel a peremes nyíláson keresztül csak akkor lehetséges, ha a sűrített tej könnyen folyó, s egyenletes állományú. A tartalmat úgy keverjük át, hogy egy mintavevő rudat a nyíláson át bevezetünk, s ameddig csak lehetséges, minden irányban mozgatjuk és forgatjuk. A rúd visszahúzása után a 3.2.1. pontban leírt módon mintát gyűjtünk.
Lehetséges megoldás még az, ha a tartály tartalmát megfelelő edénybe átöntjük, ügyelve arra, hogy az eredeti térfogatból a lehető legtöbb jusson át. A 3.2.1. pontban leírt, keverővel történő átkeverés után kivesszük a mintát.
3.2.3. Mintavétel fogyasztói csomagolásból
Sértetlen, bontatlan csomagot kell mintaként kivenni. Lehetőleg azonos gyártásból származó, azonosan megjelölt csomagokat vegyünk ki. A minta összes mennyisége legalább 200 g legyen.
3.3. A minták tartósítása, tárolása, szállítása
Lásd: Általános előírások: 5. és 6. pont.
III. 2. módszer: Mintavétel tejporból
1. Alkalmazási terület
Ez a módszer
– teljes tejpor,
– sovány tejpor,
– félzsíros tejpor,
– emelt zsírtartalmú tejpor
kémiai vizsgálatához szükséges mintavételt írja le.
2. Eszközök
Lásd: Általános előírások, 2. pont.
2.1. A szúró mintavevők legyenek elegendő hosszúak ahhoz, hogy a tartály aljára érjenek. Az engedélyezett szúró mintavevők a IV. részben leírtaknak feleljenek meg.
2.2. Kanál vagy spatula
A spatula lapja széles legyen.
2.3. Mintatároló edény
Lásd: Általános előírások, 3. pont.
3. Eljárás
3.1. Általános megjegyzések
Ügyelni kell arra, hogy a tartályban lévő termék a mintavétel alatt vagy ezt megelőzően a lehető legkevesebb légnedvességet vegye fel. A tartályt a mintavétel után gondosan vissza kell zárni.
3.2. Mintavétel kémiai vizsgálat céljára
A minta mennyisége legalább 200 g legyen. A tiszta és száraz szúró mintavevőt a terméken átfúrjuk, ehhez a tartályt megdöntjük, vagy oldalára fektetjük.
A szúró mintavevő nyílását lefelé fordítjuk, hogy a behatolás egyenletes legyen. Ha a szúró mintavevő a tartály alját elérte, 180 fokkal megfordítjuk, kihúzzuk és tartalmát a mintatároló edénybe töltjük. A 200 g-hoz egy vagy több furat szükséges. Mihelyt a szükséges mintamennyiség összegyűlt, a mintatároló edényt le kell zárni.
3.2.1. Mintavétel fogyasztói csomagolásból
Sértetlen, bontatlan csomagot kell mintaként kivenni.
Lehetőleg azonos gyártásból származó, vagy azonosan megjelölt csomagokat vegyünk ki. A minta összes mennyisége legalább 200 g legyen.
Megjegyzés: Amennyiben változó tulajdonságokat kell meghatározni, ezt a mintavételi eljárást kell alkalmazni.
3.3. A minta tárolása és szállítása
Lásd: Általános előírások, 5. és 6. pont.
IV. Szúró mintavevők csomagolatlan tejpor mintavételéhez
1. Fajták:
A típus: hosszú
B típus: rövid
(lásd: 5. ábra)
2. Eszközök
A lap és a támasz polírozott fémből, lehetőleg rozsdamentes acélból készüljön.
A hosszú szúró mintavevő fogantyúja rozsdamentes acél legyen. A rövid szúró mintavevőnek levehető, fából vagy műanyagból készült, bajonettzárral ráerősíthető nyele legyen.
3. Kivitel
3.1. Az eszköz alakja, anyaga és felülete tegye lehetővé könnyű tisztítását.
3.2. Az A típusú szúró mintavevő éle elég éles legyen ahhoz, hogy kaparóként is szolgálhasson.
3.3. A szúró mintavevő hegye legyen elég éles ahhoz, hogy a mintavételt megkönnyítse.
4. Főbb méretek
A szúró mintavevők a következő táblázatban található méreteknek feleljenek meg, 10% megengedett eltéréssel:
|
Méretek milliméterben |
|
A típus
hosszú |
B típus
rövid |
A szúró mintavevő hossza |
800 |
400 |
A fémlemez vastagsága |
1–2 |
1–2 |
A szúró mintavevő belső átmérője a hegyénél |
18 |
32 |
A szúró mintavevő belső átmérője a nyél alatt |
22 |
28 |
Nyílásszélesség a hegyénél |
4 |
20 |
Nyílásszélesség a nyél alatt |
14 |
14 |
5. A szúró mintavevők használata
5.1. A többé-kevésbé könnyen folyó porokba a szúró mintavevőt függőlegesen vezetjük be. Az A típusú szúró mintavevőt elforgatással megtöltjük és függőlegesen kihúzzuk.
A B típusú szúró mintavevőt a bevezetés során teljesen megtöltjük, és visszahúzáskor megfelelő állásban tartjuk, hogy az alsó részén az anyag kihullását elkerüljük.
5.2. A többé-kevésbé szabadon folyó porok tartályát megdöntjük, a szúró mintavevőt csaknem vízszintesen, nyílásával lefelé bevezetjük és nyílásával felfelé húzzuk ki.
1. ábra: Keverő kannához és vödörhöz
(méretek mm-ben)
2. ábra: Kisméretű tartályhoz ajánlott keverő
(méretek mm-ben)
3. ábra: A cukrozott sűrített tej keverésére alkalmas keverő 4. ábra: Mérőkanál folyadékokhoz
5. ábra: Szúró mintavevő tejporhoz
(méretek mm-ben)